Until a few years ago the polyomavirus family (Polyomaviridae) included a dozen viruses identified in avian and mammalian hosts. Two of these, the JC and BK-polyomaviruses isolated a long time ago, are known to infect humans and cause severe illness in immunocompromised hosts. Since 2007 an unprecedented number of eight novel polyomaviruses were discovered in humans. Among them are the KI- and WU-polyomaviruses identified in respiratory samples, the Merkel cell polyomavirus found in skin carcinomas and the polyomavirus associated with trichodysplasia spinulosa, a skin disease of transplant patients. Another four novel human polyomaviruses were identified, HPyV6, HPyV7, HPyV9 and the Malawi polyomavirus, so far not associated with any disease. In the same period several novel mammalian polyomaviruses were described. This review summarizes the recent developments in studying the novel human polyomaviruses, and touches upon several aspects of polyomavirus virology, pathogenicity, epidemiology and phylogeny.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
The polyomaviruses have been recognized as a separate virus family (Polyomaviridae) consisting of several species since 1999. Before that time they formed the genus Polyomavirus in the family Papovaviridae that contained the papillomaviruses, polyomaviruses and the simian vacuolating agent 40 (SV40). Nowadays, the latter virus forms the namesake species that is designated the polyomavirus type species (prototype) as listed in the ICTV 9th Report (Norkinet al., 2012).
The first members of the polyomavirus family, mouse polyomavirus (MPyV) and SV40, were identified halfway through the last century as filterable agents that caused tumours in newborn mice and hamsters (Eddy et al., 1962; Girardi et al., 1962; Gross, 1953). SV40 contaminated many lots of human poliovirus vaccine produced in monkey kidney cells. This raised a serious concern, but so far SV40 has never been shown to be oncogenic in humans (Strickler et al., 1998). The first two human polyomaviruses were discovered in 1971. The JC-polyomavirus (JCPyV) was identified in a brain tissue extract from a progressive multifocal leukoencephalopathy (PML) patient that bore the initials J. C. (Padgett et al., 1971). The first BK-polyomavirus (BKPyV) reported was isolated from the urine of a nephropathic kidney transplant patient B. K. (Gardner et al., 1971). Both JCPyV and BKPyV were not oncogenic in humans despite being closely related phylogenetically with SV40 (Johne et al., 2011). Subsequently, more polyomaviruses were identified in rodents, cattle and birds, but not in humans until this century.
From 2005 on, eight novel human polyomaviruses (HPyV) were identified. Two of those are involved in disease development in immunosuppressed and/or elderly patients, and therefore seem to fit the ‘opportunistic’ polyomavirus profile characterized by symptomatic reactivations after a period of persistent latent infection. For the other novel HPyVs pathogenicity is still unknown. In this review, the novel HPyVs will be discussed with respect to virological and pathogenic properties as far as they are known, addressing some epidemiological and phylogenetic aspects of polyomavirus in general.
Fig. 1 shows a timeline of polyomavirus (putative) species identifications at the time of writing (October 2012), animal and human. A list of polyomaviruses that prototype the (tentative) namesake species, name abbreviations and RefSeq/GenBank numbers of genome sequences are shown in Table 1. In this review, the polyomaviruses will be termed according to their species names and abbreviations mentioned in the pending 2011 ICTV taxonomy proposal regarding the family Polyomaviridae, as presented in a recent publication on the subject (Johne et al., 2011). The phylogenic tree composed by Johne, Norkin and collaborators is shown in Fig. 2. The most recent polyomavirus discoveries are not mentioned in the pending ICTV proposal. These will be termed according to their provisional names given in their first publication, respectively.
Fig. 1.
A time line that indicates 32 (putative) polyomavirus species discovered at the time of writing (October 2012). The year of discovery refers to the first description of the virus, not necessarily to the year that the full genome DNA sequence was obtained. The virus name abbreviations are explained in Table 1where also the citations to the first descriptions can be found.
Table 1. List of 32 polyomavirus names identified up to October 2012, which prototype putative species
Fig. 2.
Phylogenetic tree of 25 polyomaviruses based on whole genomic nucleotide sequences, as published before by Johne, Norkin and others (Johne et al., 2011).
New human polyomaviruses and methods of discovery
With the steady advancement of nucleic acid amplification and detection techniques, the need for cytopathic changes observed in cell culture to detect the presence of a virus was bypassed. Recent versions of these molecular techniques allow sensitive and high-throughput analyses of large numbers of clinical samples that led to a revolution in virus discovery. Nucleic acid isolated from both diseased and healthy tissues can be used as ‘a template’ for such techniques and therefore the identified viruses are not necessarily pathogenic. These techniques are frequently combined with strategies to enrich the original sample for viral DNA or RNA, by reducing the content of host genomic DNA.
KI-polyomavirus (KIPyV) and WU-polyomavirus (WUPyV)
In 2007 almost coincidentally, two novel HPyV were reported, the Karolinska Institute polyomavirus (KIPyV) identified in Stockholm, Sweden and the Washington University polyomavirus (WUPyV) identified in St Louis, USA. Both were discovered in respiratory tract samples from individuals with (acute) respiratory tract infections, but by totally different methods.
KIPyV was identified by pooling centrifuged and filtered supernatants from randomly selected DNase-treated nasopharyngeal aspirates, from which DNA and RNA were extracted (Allander et al., 2007). The nucleic acids served as a template for random-PCR, and the products were separated by gel electrophoresis, cloned and sequenced. This resulted in several sequence reads that were subsequently trimmed, clustered and sorted using dedicated software. Finally, BLAST searches were performed to look for similarities with known (viral) sequences listed in GenBank. Details of the technical approach were published in a previous virus discovery paper describing the identification of the human bocavirus (Allander et al., 2005).
For the identification of WUPyV a ‘shotgun sequencing’ strategy was used on a single nasopharyngeal aspirate that proved negative for 17 known viral respiratory pathogens in diagnostic PCR. Total nucleic acid isolated from the sample was randomly amplified and PCR products were cloned and sequenced. The reads were analysed in a comparable fashion to the above, which revealed several sequences that displayed significant homology with JCPyV and BKPyV (Gaynor et al., 2007). Subsequent analyses of the compiled WUPyV genome showed highest sequence identity with the KI-polyomavirus just isolated from respiratory samples, described above.
Merkel cell polyomavirus (MCPyV)
Exciting and worrisome at the same time has been the discovery of the MCPyV, which finally proved assumptions of polyomavirus involvement in human oncogenesis true. Merkel cell carcinomas (MCC) are rare but aggressive cutaneous tumours of neuroendocrine origin that typically develop in the elderly and immunocompromised patients. Using a technique called digital transcriptome subtraction (Feng et al., 2007), MCPyV was identified in MCC (Feng et al., 2008). This technique compares (subtracts) cDNA libraries generated with random RT-PCR from diseased and healthy tissues and thereby identifies mRNA transcripts potentially unique for either or both. In this case with the help of sophisticated software, unique sequences were further analysed and some of them displayed homology to the monkey B-lymphotrophic polyomavirus (LPyV) and BKPyV. Through primer-genome-walking the complete genome was sequenced. New to HPyVs, but known for oncogenic animal polyomaviruses detected in tumours, the MCPyV genome was found integrated into the host genome (Feng et al., 2008).
Trichodysplasia spinulosa-associated polyomavirus (TSPyV)
Trichodysplasia spinulosa is a rare follicular skin disease of immunocompromised patients that will be described further below. In 1999, Haycox and coworkers proposed a viral origin, because affected hair follicle cells contained clusters of virus particles (Haycox et al., 1999). It took until 2010 before the identity of this virus was uncovered (van der Meijden et al., 2010), with the help of a technique called rolling-circle amplification (RCA) (Johne et al., 2009). This technique exploits random hexamer primers and a DNA polymerase (Phi-29) with proof-reading and strand-displacement properties. If a sample contains a relative excess of a particular small circular dsDNA molecule, for instance a plasmid or a (small) viral genome, RCA will produce long, high-molecular DNA molecules with concatenated (linearized) stretches of DNA of potential interest. These stretches will appear as sharp bands on gel when the RCA-product is cut with specific restriction enzymes and can be cloned and sequenced subsequently. This approach already proved successful in the past to identify a number of animal papillomaviruses and polyomaviruses and finally revealed the genome of TSPyV (Johne et al., 2009;van der Meijden et al., 2010).
Human polyomaviruses 6 and 7 (HPyV6 and HPyV7)
In order to detect free, non-integrated, MCPyV genomes on the skin, RCA was performed on forehead swab samples of healthy individuals. With some adjustments to increase RCA sensitivity, such as DNA exonuclease treatment to digest cellular (linear) DNA, this approach revealed the identity of a new polyomavirus, HPyV6, on the skin of a healthy individual (Schowalter et al., 2010). With the use of a new degenerate PCR primer set to detect HPyV6- and WUPyV-like sequences, an additional novel polyomavirus was found in these healthy skin samples that was called HPyV7 (Schowalter et al., 2010).
Human polyomaviruses 9 (HPyV9)
Several serological studies already showed the presence of ‘cross-reactive’ antibodies in a percentage of human sera that recognized LPyV (Kean et al., 2009; Viscidi & Clayman, 2006), which was isolated from an African green monkey (Pawlita et al., 1985). Furthermore, LPyV-derived sequences were obtained from human blood, indicating the occurrence of LPyV(-like) infections in humans (Delbue et al., 2008). In 2011, a novel polyomavirus which closely resembled LPyV was identified independently by two research groups in human serum and skin swabs, respectively, and tentatively called HPyV9. For screening of the serum samples a degenerate polyomavirus PCR primer set was used (Leendertz et al., 2011), which generated an unknown, putative HPyV sequence next to many known other HPyV sequences. With primer-walking the rest of the HPyV9 sequence was uncovered (Scuda et al., 2011). In the other report, DNA isolated from healthy skin swabs was subjected to high-throughput sequencing, which resulted in millions of reads and several contigs that matched MCPyV, HPyV6 and HPyV7, as well as many human papillomaviruses. Some contigs showed the highest homology with LPyV and after filling the contig sequence gaps by designing additional primers, a virus genome was found that coincided with HPyV9 (Sauvage et al., 2011).
Malawi polyomavirus (MWPyV/HPyV10)
Recently, another novel human polyomavirus was identified in stool of a 1-year-old healthy child from Malawi (Siebrasse et al., 2012). This virus was discovered by suspending a small amount of faeces that subsequently was centrifuged and filtered through 0.45 and 0.22 µm pores. After chloroform extraction-free-DNA was removed by DNase treatment. The solution that still contained encapsidated DNA was SDS and Proteinase K-treated, and DNA was isolated. Finally, this DNA was subjected to RCA and pyrosequencing. Hundreds of reads were obtained, sequenced and aligned to sequences within the GenBank database. This revealed several polyomavirus-like reads and with the use of a primer walking strategy the complete MWPyV genome was fulfilled (Siebrasse et al., 2012). Very recently an almost identical virus called HPyV10 was identified in an anal wart of an immunocompromised patient with WHIM syndrome with the help of RCA (Buck et al., 2012). MWPyV and HPyV10 probably represent a single species.
Genome and (putative) proteins
Genome organization and transcription
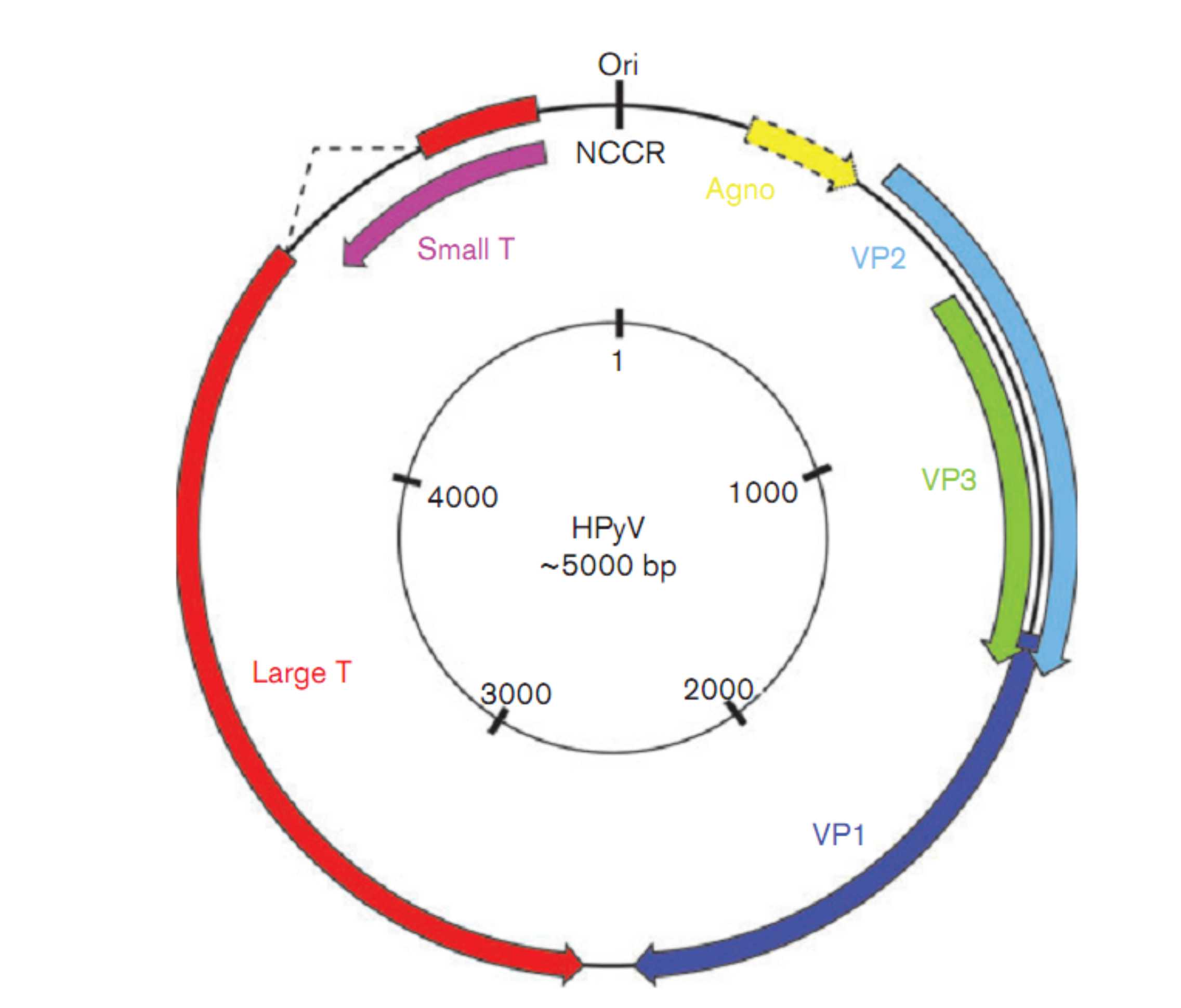
Viruses of the family Polyomaviridae, including the recently identified human viruses, are among the smallest known to infect humans, in relation to both particle size and genome length (Imperiale & Major, 2007). The 45 nm non-enveloped virion harbours a circular dsDNA genome of approximately 5000 bp that is divided into three regions, the non-coding control region (NCCR) and the early and late coding regions (Fig. 3).
Fig. 3.
Schematic representation of a human polyomavirus genome made up of circular dsDNA. Regions that code for the indicated gene products are shown in colour. The agnoprotein-encoding gene is not found in every HPyV genome.
The NCCR contains the origin of replication flanked by several large T-antigen-binding sites, as well as the transcription promoters and regulatory elements. The novel human polyomaviruses display a similar NCCR organization. In JCPyV- or BKPyV-diseased individuals with viral DNA detectable in different body compartments, NCCRs of these isolates can vary from what is called the ‘archetype’ NCCR that is thought to be the NCCR found in transmissible virus found in urine. It is believed that the NCCR mutations, deletions and repeats increase T-antigen transcription and the replication rate, as was shown for instance for JCPyV (Gosert et al., 2010). Systematic mutual analyses of the NCCR regions among different isolates within individuals have not been reported yet for the novel human polyomaviruses.
The early coding region contains the genes for the small and large T-antigens that are transcribed through alternative splicing. The late coding region found on the opposite strand codes for the VP1, VP2 and VP3 structural capsid proteins (Fig. 3). At the 3′ ends, the early and late regions are separated by a small intergenic region. Analogous to JCPyV and BKPyV, so far no ORFs have been found in the novel human polyomaviruses to indicate the presence of a middle T-antigen as found in rodent polyomaviruses. The agnoprotein and VP4 expressed by JCPyV, BKPyV and several animal polyomaviruses including birds seem not to be encoded by the novel HPyVs (Johne & Müller, 2007), but for most of the novel viruses the transcription-patterns have not been revealed yet. In addition to a number of transcripts coding for the complete small and large T-antigens, MCPyV and SV40 may use a 17kT-like transcript called T4 or 57kT that directs the synthesis of large T-antigen with a large central splice (Shudaet al., 2008; Zerrahn et al., 1993).
Besides transcription regulation by the NCCR, T-antigen expression was shown to be modulated post-transcriptionally by microRNAs in SV40 (Sullivan et al., 2005). For JCPyV and BKPyV a viral microRNA was described that targets the stress-induced ligand ULBP3 and thereby inhibits (antiviral) NK T-cell activity (Bauman et al., 2011). For MCPyV, a microRNA was detected that potentially regulates early gene expression (Seo et al., 2009). Moreover in MCC, MCPyV-encoded microRNAs were identified that potentially regulate T- and B-cell receptor signalling hampering viral (tumour) immune recognition (Lee et al., 2011).
T-antigens
The T-antigens play essential roles in viral transcription and replication, as well as in host cell tuning to enable virus replication (Imperiale & Major, 2007;Sullivan & Pipas, 2002). As polyomaviruses are completely dependent on the host cell replication machinery, a significant part of this tuning is aimed at inducing S-phase and bypassing cell cycle-control measures. In some cases, these features can result in uncontrolled cell growth and eventually tumour formation. So far, MCPyV is the only (new) HPyV for which these phenomena have been noted (Chang & Moore, 2012). As a consequence, the T-antigens of MCPyV have been studied already quite extensively (see below, and the MCPyV Pathogenicity paragraph), whereas for the other new HPyVs this knowledge is almost entirely based on nucleotide and predicted amino acid sequence properties.
The small and large T-antigens share their N terminus, while having C-terminal regions of different sizes encoded in different reading frames that are expressed by alternative splicing. The novel human polyomaviruses seem no exception to this rule. This shared region occupied by the so-called J-domain contains important motifs for virus replication and cellular transformation, such as CR1 and DnaJ (Sullivan & Pipas, 2002). Downstream, the small T-antigen contains a PP2A subunit-binding motif probably to activate the Akt-mTOR pathway related to (tumour) cell survival (Andrabi et al., 2011; Buchkovich et al., 2008). The large T-antigen further includes a number of functionally important sequence signatures: a nuclear-localization signal, an origin-binding domain and an ATPase domain-containing helicase domain (Arora et al., 2012;Sullivan & Pipas, 2002), as well as a pRB-binding motif and p53-interaction domain that probably play a role in cellular transformation. Apart from MCPyV (Shuda et al., 2008), the presence of (most of) these motifs was identified by bioinformatics in TSPyV and HPyV9 (Scuda et al., 2011; van der Meijden et al., 2010), but they remain to be characterized functionally.
VP capsid proteins
The late VP1, VP2 and VP3 structural proteins together make up the 40–45 nm polyomavirus capsid (Baker et al., 1989). Five VP1 molecules form a capsomer that interacts with either a VP2 or VP3 molecule to autoassemble into a capsid composed of 72 VP1 pentamers (Imperiale & Major, 2007). Similar sequences probably encoding VP1, VP2 and VP3 have been identified in all of the novel polyomaviruses. So far there is no reason to believe that the novel viruses will behave differently with respect to capsid formation, although there is debate whether MCPyV VP3 is functional (Pastrana et al., 2009). VP4, noted for SV40 and expressed very late in infection probably serving as a porin in the cellular membrane facilitating viral release (Daniels et al., 2007; Raghava et al., 2011), has not been reported or predicted for any of the novel HPyVs.
How the novel polyomaviruses recognize and infect their host target cells is largely unknown. Most of the known polyomaviruses interact by linkage of the major capsid protein VP1 with terminal sialic acids (alpha2,3 or alpha2,6) of mainly gangliosides on the host cell membrane (Neu et al., 2009), after which they are internalized using different entry pathways. For MCPyV, it was recently shown that this virus uses glycosaminoglycans for initial attachment, whereas later in the encounter sialic acids may still be important (Schowalter et al., 2011). Future research has to reveal whether the other novel human polyomaviruses use similar strategies for host cell recognition.
Virus pathogenicity, associated diseases and clinical aspects
For two of the novel HPyVs a causal relationship with a specific clinical condition is highly probable. In both cases the disease/cancer has lent its name to the virus, trichodysplasia spinulosa to TSPyV and MCC to MCPyV. For MCPyV, an additional association with chronic lymphocytic leukaemia is suggested, and for TSPyV an association has been suspected with pilomatricoma. Both will be addressed here as well. For KIPyV and WUPyV, the evidence for involvement in respiratory tract infections is weak and for the other human polyomaviruses such associations are absent to date.
MCPyV and MCC
MCC is a rare aggressive neuro-endocrine tumour of epidermal origin with an increasing incidence of approximately 0.2–0.5 per 100 000 depending on age, skin type, (cumulative) sun exposure and immune status (Schrama et al., 2012;Van Keymeulen et al., 2009). The association with fair skin and sun exposure suggests a direct effect of UV-radiation on MCC development, but this has not been confirmed experimentally. In 2008, the presence of MCPyV in MCC was shown for the first time (Feng et al., 2008), a finding which has been confirmed by many researchers worldwide showing that at least 75 % of all MCC are MCPyV-positive (Arora et al., 2012).
Several findings indicate direct involvement of MCPyV in Merkel cell carcinogenesis. For instance, in almost all cancer cases MCPyV is found linearized and clonally integrated in the host cell genome, although the site of integration appears random (Feng et al., 2008; Laude et al., 2010; Martel-Jantinet al., 2012). Furthermore, the MCPyV large T-gene was found to carry ‘signature’ mutations specific for the carcinomas. Their expression results in a truncated large T-protein that lacks the C-terminal helicase domain, the putative p53-interaction domain and, sometimes, also the origin-binding domain (Feng et al., 2008; Sastre-Garau et al., 2009; Shuda et al., 2008). As a consequence, the function of large T in viral replication is selectively impaired. Small T-antigen is unaffected by these mutations. So in an apparent parallel to other virus-associated cancers, e.g. human papillomavirus (HPV)-associated cervical cancer, also in MCPyV-positive MCC there seems to be a strong association with non-productive MCPyV infection. Probably the strongest evidence of pathogenicity (oncogenicity) of MCPyV is provided by studies showing that both large and small T-antigen expression are indispensable to maintain cellular transformation. If T-antigen expression was inhibited experimentally by short interfering (hairpin) RNAs, MCC cell lines underwent growth arrest or died (Houben et al., 2010; Shuda et al., 2011).
The presence of MCPyV-negative (20–25 %) and MCPyV-positive (75–80 %) MCC underscores the existence of at least two genetic forms of this cancer, possibly with a different prognosis and different demands for treatment. When comparing the prognosis of MCPyV-positive and -negative MCC, there seems to be a trend that MCPyV-positive ones do better and carry less genomic mutations, for instance in TP53, than the MCPyV-negative ones (Bhatia et al., 2010; Sihto et al., 2009, 2011). Serological studies point in the same direction (see below). Other studies, however, report no difference in clinical outcome between MCPyV-positive and -negative cancers (Hall et al., 2012; Schrama et al., 2011), underscoring the necessity of further study in this regard. Also, the role of MCPyV-directed cellular immunity in MCC development and progression, as well as tumour immune-escape needs further clarification (Iyeret al., 2011; Kumar et al., 2011).
MCPyV and chronic lymphocytic leukaemia
It has been noted for some time that MCC, although rare, are more frequently seen in patients with chronic lymphocytic leukaemia (CLL) and vice versa, suggesting a common nature (Howard et al., 2006; Kaae et al., 2010; Tadmoret al., 2011). Although very few MCC from CLL patients have been analysed, all of them harboured MCPyV (Koljonen et al., 2009; Tolstov et al., 2010). Based on this information and the putative lymphotropism of MCPyV (Toracchio et al., 2010), studies were conducted to look for MCPyV in peripheral blood mononuclear cells of CLL patients. The first study in that regard could detect MCPyV DNA and T-antigen in a small minority of the CLL cases (Shuda et al., 2009; Tolstov et al., 2010). The calculated viral loads, however, were considered too low to suspect a significant contribution of MCPyV to the CLL phenotype.
Other groups have detected MCPyV DNA in 25–35 % of CLL samples (Imajoh et al., 2012; Pantulu et al., 2010; Teman et al., 2011). Also in these cases, the measured viral loads were several logs lower than the ~1 copy per cell shown for the MCC, thereby excluding viral involvement in maintenance of the transformed tumour state. Nevertheless, a third of the MCPyV-positive samples carried a specific large T-antigen mutation potentially blocking the viral helicase activity and suggestive of a role for the virus in oncogenesis (Pantuluet al., 2010). Integration of the virus in CLL was not demonstrated, but fluorescence in situ hybridization patterns of MCPyV-positive samples were suggestive in this regard (Haugg et al., 2011). The finding that MCPyV DNA is also present in buffy coats from healthy blood donors further obscures the issue of MCPyV involvement in CLL (Pancaldi et al., 2011).
TSPyV and trichodysplasia spinulosa
Trichodysplasia spinulosa is a rare skin disease exclusively found in severely immunocompromised hosts, especially solid organ transplant recipients (Kazem et al., 2012; Matthews et al., 2011). It is characterized by gradual development of papules and spicules (spines) on the face, sometimes accompanied by alopecia of the eyebrows and lashes (Wanat et al., 2012). Worldwide some 30 cases have been described, the first as a cutaneous side effect of cyclosporin-A use (Chastain & Millikan, 2000; Izakovic et al., 1995). Haycox and coworkers proposed for the first time that trichodysplasia spinulosa concerns a viral disease and showed the presence of intranuclear clusters of honeycomb-arranged viral particles in the distended and dismorphic hair follicles, especially in proliferating inner root sheath cells that over expressed trichohyalin (Haycox et al., 1999). Subsequent reports confirmed the typical histological findings of hyperplastic hair bulbs and the presence of polyomavirus or papillomavirus-like particles (Lee et al., 2008; Osswald et al., 2007; Sadler et al., 2007; Sperling et al., 2004; Wyatt et al., 2005). Attempts to identify the nature of this virus failed until TSPyV was identified with the help of RCA in 2010 (van der Meijden et al., 2010).
To date the presence of TSPyV in trichodysplasia spinulosa lesions has been confirmed in patients from Western Europe, North America and Australia (Fischer et al., 2012; Kazem et al., 2012; Matthews et al., 2011; Wanat et al., 2012). In a small case-control study, we showed the presence of TSPyV DNA in all of the tested trichodysplasia lesions and only in a small number of healthy control samples (P<0.001) (Kazem et al., 2012). TSPyV DNA loads measured in these lesions were at least 4 logs higher those in healthy skin(-derived) tissues. Furthermore, we showed that TSPyV VP1-antigen was detected only in the affected hair bulbs, confined to trichohyalin-overexpressing inner root sheath cells. Taken together, these data indicate an aetiological relationship between active TSPyV infection and trichodysplasia spinulosa. In one patient for whom this analysis was performed, a decline in TSPyV load was observed with successful antiviral (cidofovir) treatment of trichodysplasia spinulosa (van der Meijden et al., 2010). Whether trichodysplasia spinulosa concerns a symptomatic reactivation of a persistent TSPyV infection or a symptomatic primary TSPyV infection in the window of (very) poor immunity is unclear at the moment. The rarity of the condition in the background of the high seroprevalence in the general population seems to favour the latter possibility, as primary infections at higher age may be considered a relatively rare event. In both scenarios, next to immunity, additional (host) factors are likely to influence the occurrence of clinically relevant TSPyV-infection.
TSPyV and pilomatricoma
Because of the tropism of TSPyV for the hair follicle area, and the oncogenic properties of some other polyomaviruses, a relation was suspected between TSPyV infection and pilomatricoma, a benign skin tumour originating from proliferating hair-matrix cells. Previously, it was shown that the murine MPyV induces hair follicle tumours in mice (Dawe et al., 1987; Freund et al., 1992). Using a rabbit antiserum raised against MPyV VP1 (Leavitt et al., 1985), 40 % of tested human pilomatricomas stained positive (Sanjuán et al., 2010). Attempts to detect TSPyV in these tumours however failed; both TSPyV DNA and VP1-antigen could not be detected in ten pilomatricoma samples (Kanitakis et al., 2011). So, despite experimental evidence that shows polyomavirus tropism for the hair follicle cells, no correlation between TSPyV and pilomatricoma could be established.
KIPyV/WUPyV and respiratory tract infections
Both KIPyV and WUPyV were identified in respiratory samples. Ever since, several researchers have tried to establish an association between upper and/or lower respiratory tract infections and the presence of these viruses. To summarize, both viruses were regularly detected in respiratory materials (Bialasiewicz et al., 2008), but none of these studies revealed a clear correlation between KIPyV and WUPyV and respiratory disease (Babakir-Mina et al., 2011). Often KIPyV and WUPyV were detected together with other known (viral) pathogens and not in convincingly high loads to suspect a causal relationship. The influence of host immune status on this possible association is also not clear at the moment (Csoma et al., 2011; Rao et al., 2011).
HPyV6, HPyV7, HPyV9 and MWPyV/HPyV10
So far, for HPyV6, HPyV7 and HPyV9 no disease association was reported or suggested. Future studies should resolve whether MWPyV/HPyV10 can be considered a cutaneotropic virus contributing to anal wart development, as suggested in the WHIM patient (Buck et al., 2012), or rather as a faecally transmitted pathogen or contaminant (Siebrasse et al., 2012).
Serology and seroprevalence
Cross-sectional studies of the known human polyomaviruses JCPyV and BKPyV have shown that both infections detected via the presence of VP1-directed seroresponses are prevalent in the general population. Depending on the study, the seroprevalence of JCPyV is approximately 50 % and BKPyV reaches almost 90 % (Kean et al., 2009). Serological studies of the novel human polyomaviruses revealed a considerable variation in the presence of serum antibodies (Carter et al., 2009; Chen et al., 2011b; Kantola et al., 2010; Kean et al., 2009; Schowalteret al., 2010; Touzé et al., 2010; Trusch et al., 2012; van der Meijden et al., 2011; Viscidi et al., 2011). The lowest seroprevalence was detected for HPyV9 varying between 15 and 45 % (Nicol et al., 2012; Trusch et al., 2012; E. Van der Meijden and M. C. W. Feltkamp, unpublished results), the highest for WUPyV, TSPyV and HPyV6 60–95 % (Chen et al., 2011b; Kean et al., 2009; Nguyen et al., 2009; Schowalter et al., 2010; van der Meijden et al., 2011). In all serological studies, the strongest increase in seroprevalence is observed at a young age, indicating that primary infections generally occur in (early) childhood (Chen et al., 2011a, b; Kean et al., 2009; van der Meijden et al., 2011; Viscidi et al., 2011). So far, the seroprevalences of the novel HPyVs measured with VLPs and GST–VP1 fusion proteins appear similar, as was shown for instance for TSPyV (Chen et al., 2011b; van der Meijden et al., 2011).
For MCPyV, case-control studies were performed to investigate the relation between MCPyV-specific seroresponses and MCC. In all reports, statistically significant associations were found between the VP1-directed seroresponses, measured against VP1–GST-fusion proteins and VLPs, and MCC (Carter et al., 2009; Pastrana et al., 2009; Touzé et al., 2011). Furthermore, high-titre MCPyV VP1 responses were almost exclusively found in the carcinoma cases and correlated with better survival (Pastrana et al., 2009; Touzé et al., 2011). The best correlation for MCPyV serology and MCC were obtained when the T-antigen is used as the target antigen. Seroresponses against the T-antigens are virtually absent in healthy individuals, whereas almost half of the MCC patients responded to this antigen (Paulson et al., 2010). Moreover, a T-antigen titre rise was correlated with progressive disease and a decline with remission.
Polyomavirus persistence and reactivation and spread of infection
Persistence and reactivation
It is generally believed that primary infection of human polyomaviruses is followed by a persistent, asymptomatic (latent) infection with very low levels of replication that remains in the body lifelong. For JCPyV and BKPyV reactivation from the latent state and manifestation of symptomatic infection is observed only in the case of reduced immunity, for instance in long-term immunosuppressed solid organ transplant recipients (BKPyV, nephropathy) and in AIDS patients (JCPyV, PML) (Imperiale & Major, 2007). Nowadays JCPyV-caused PML is increasingly observed in Natalizumab-treated patients, which are not overall immunocompromised, but carry targeted adhesion molecules that prevent T-lymphocyte homing, for instance to the brain (Bloomgren et al., 2012; Major, 2010).
In healthy individuals, the detection rate of JCPyV and BKPyV DNA is generally much lower than their seroprevalence. In urine of healthy blood donors for example, the detection rate of JCPyV and BKPyV DNA was approximately 3- and 10-fold lower than the seroprevalence measured within the same group, respectively (Egli et al., 2009). In immunosuppressed solid organ transplant patients and haematopoietic stem cell recipients, however, BKPyV can be found in urine in the majority of (seropositive) cases, often in very high amounts indicative of massive reactivation of infection (Bennett et al., 2012; Hirsch et al., 2009).
For the novel HPyVs the association between diminished immunity and reactivation is less clear. Only for TSPyV, with a seroprevalence of 70 % (Chen et al., 2011b; van der Meijden et al., 2011), are clinical consequences exclusively seen in severely immunocompromised hosts (Kazem et al., 2012; Matthews et al., 2011). However, the TSPyV DNA detection rate, on the skin at least, hardly varies among immunocompetents (2 %) and immunocompromised hosts (4 %) (Kazem et al., 2012; van der Meijden et al., 2010). For (MCPyV-positive) MCC, a weaker association with immunosuppression is found, and also the elderly are at increased risk of developing this tumour, possibly explained by immunosenescence. Also for MCPyV, the DNA detection rates on the skin do not differ much between immunocompetents and immunocompromised, albeit they are generally much higher than for TSPyV and resemble the seroprevalence of 50 % (Wieland et al., 2011). In general, it should be noted that both MCC and trichodysplasia spinulosa are very rare conditions implying that (host) factors other than immunity also play a role in controlling infections of these viruses. For KIPyV, WUPyV and HPyV6, HPyV7, HPyV9 and MWPyV the relation with immunity is either not known or poorly described at the moment. This is largely explained by the lack of an established clinical condition that could be attributed to one of these novel viruses and the unawareness of the organ where these viruses latently persist.
Spread of infection
From studies of JCPyV and BKPyV it is known that they are found in urine, faeces and saliva. The excreted numbers of virus are so high that these polyomaviruses even serve as markers of sewage pollution of for instance recreational waters (Korajkic et al., 2011). Which route of virus excretion primarily drives human infection is not precisely known, but the efficiency of infection seems clear from the high seroprevalence numbers, for both the known and novel human polyomaviruses.
Regarding possible routes of excretion and transmission of cutaneous MCPyV, a recent study showed the presence of viral DNA on 75 % of samples from environmental surfaces (door handles, ticket machines etc.). DNase sample treatment and viral load measurement suggested that about 5 % of the detected MCPyV DNA was protected, probably encapsidated and therefore potentially infectious (Foulongne et al., 2011). TSPyV DNA has been occasionally detected in urine and kidney tissue (Fischer et al., 2012; Scuda et al., 2011), but if this provides a clue regarding transmission remains to be seen.
Phylogeny and evolutionary trends: how many HPyVs are around?
The ten known HPyVs comprise the largest host-specific subset of the polyomaviruses described so far. This probably reflects a bias of the virus discovery effort that increasingly focuses on hunting for human viruses, particularly during the past several years as was detailed above. Pairwise divergence between HPyVs varies as much as in the entire polyomavirus dataset. If measured for the combined VP1, VP2 and LT proteins it ranges from 15 (for BKPyV/JCPyV pair) to 61 % (BKPyV/HPyV6), or, if repeated substitutions accounted, from 0.17 (BKPyV/JCPyV) to 1.52 (TSPyV/HPyV6) substitutions per position on average (Fig. 4). Apart from BKPyV/JCPyV, also the KIPyV/WUPyV and HPyV6/HPyV7 pairs are among the most closely related, which correlates with putative shared tropism for the respiratory tract and the skin, respectively.
Fig. 4.
Genetic comparison between ten known human polyomaviruses. Two measures of genetic distance are shown for the combined polyomavirus protein set comprising VP1, VP2 and LT. The upper-right triangle shows uncorrected dissimilarity percentages (1 corresponds to 100 % dissimilarity; 0 means identical sequences); the lower-left triangle shows evolutionary distances that correct for multiple substitutions at the same site (the scale is mean substitutions per sequence position) under the WAG amino acid substitution model and rate heterogeneity among sites.
In Fig. 5 a novel tentative polyomavirus phylogenetic tree is shown. In addition to the Johne tree in which the polyomavirus genera have been proposed (Fig. 2) (Johne et al., 2011), the new tree includes seven animal and human viruses, among which HPyV9 and MWPyV/HPyV10, that represent additional putative polyomavirus species. From Fig. 5 it is clear that the HPyVs do not form a monophyletic cluster. Rather they are distributed unevenly among four out of five distant lineages. Two lineages that include human polyomaviruses belong to the genus Orthopolyomavirus (Johne et al. 2011), and are called Orthopolyomavirus-I and -II, respectively, for the sake of discussion. The third lineage belongs to the genus Wukipolyomavirus and the fourth comprises MWPyV alone.
Fig. 5.
Unrooted phylogenetic tree of 32 (putative) polyomavirus species based on LT, VP1 and VP2 protein comparisons. The tree is based on the alignment of concatenated VP1, VP2 and LT amino acid sequences. Regarding viruses found in both datasets, the obtained branching pattern (topology) of basal nodes in the tree matches that proposed by Johne and colleagues (Johne et al., 2011). One distinct clade designated the Avipolyomaviruses contains only the bird PyV types. The Wukipolyomavirus lineage supports four HPyV types (shown in red), the Malawipolyomavirus lineage only one. Other HPyVs are clustered in the proposed Orthopolyomavirus-I and -II lineages (see text). Bar indicates number of substitutions per site. Numbers at branching events represent probability support values ranging from 0 (no support) to 1 (best support). Only probability support values lower than 1 are shown. The full polyomavirus name can be found inTable 1.
The Malawipolyomavirus and Wukipolyomavirus lineages with one and four viruses, respectively, are least populated and include exclusively HPyVs. In contrast, each Orthopolyomavirus-I and -II include 11 putative species from which only two and three, respectively, are composed of viruses that seem to infect humans. The fifth lineage, comprising no HPyVs, belongs to the genusAvipolyomavirus.
Because of the observed phyletic distribution of HPyVs among different lineages, it may be envisaged that a number of human polyomaviruses emerged as result of cross-species jumps of zoonotic viruses. For instance, TSPyV might have originated from an ancestral virus of orangutans that harbour the closely related OraPyV1 polyomavirus and MCPyV from a virus of gorillas, host to the related GggPyV virus (Fig. 5). Since the genetic diversity accommodated by the genus Wukipolyomavirus is expected to be on a par with those in the Orthopolyomavirus-I and -II clusters, the former genus is also likely to include non-human viruses that either are yet to be discovered or have gone extinct. Alternatively, polyomaviruses might have diverged by virus–host co-evolution, which was the most popular model of evolution for these viruses until a few years ago when novel human polyomaviruses started to come to light (Shadan & Villarreal, 1995; Pérez-Losada et al., 2006). Recently, the applicability of this model to two species of best-sampled human viruses, BKPyV and JCPyV (Shackelton et al., 2006; Krumbholz et al., 2008; Zheng et al., 2007), as well as the entire family (Krumbholz et al., 2009) was questioned. Particularly, the intrahost substitution rate of the two human polyomaviruses was revised by approximately two orders of magnitude upward from that expected upon the divergence by virus–host co-evolution (Chen et al., 2004;Firth et al., 2010; Shackelton et al., 2006). Consequently, polyomaviruses may evolve at rates approaching those of RNA viruses. This should be taken into account when considering the above scenarios of virus speciation, withstanding the lack of reliable estimates of interhost substitution rates.
Resolving the uncertainty about the origins of human polyomaviruses will require extending the virus discovery effort. For example, continuing the hunt for human viruses and characterizing of other hosts should lead to considerable improvement in our understanding of the evolution of polyomaviruses. Regardless of the future progress with virus discovery, the presence of five major lineages in the polyomavirus tree calls for further refinement of the recently revised taxonomy of polyomaviruses to better accommodate their diversity (Fig. 5).
Concluding remarks
With the availability of sensitive molecular techniques to detect unknown genome sequences in the background of human DNA, the discoveries of polyomaviruses have increased significantly over the past few years. It is expected that these discoveries will continue in the coming years. Whether the scale of these discoveries will reach HPV-like proportions with tens of novel (geno)types being identified over the past few years (Bernard et al., 2010;Forslund, 2007) remains to be seen.
Regarding pathogenicity, the majority of the novel HPyVs are so far without a (possibly) associated disease. More studies are needed to identify and correlate the presence of the novel viral sequences with specific diseases. Because of the nature of polyomaviruses, next to diseases caused by acute infection, these studies will probably focus on chronic diseases and cancer as well. Furthermore, as some of the novel HPyV-associated diseases are not exclusively seen in severely immunocompromised hosts, immunocompetent subjects may be regarded as well. In general, more research is needed to understand the role of immunosurveillance in controlling the novel HPyVs and the role of genetic host factors in this regard.
To study pathogenic mechanisms of the HPyVs in more detail, more knowledge should be obtained about the tropism of each virus. Discrepancies between detection rates and seroprevalence rates, in both immunosuppressed and immunocompetent hosts, suggest that at least some of the newly identified HPyVs persistently infect the host in a niche that is not yet identified or sampled properly. Knowledge about HPyV tropism and reservoir is indispensable to identify the cell type(s) needed for further cellular and molecular studies, which can guide research strategies to further understand and cope with these viruses.
- Mariet C. W. Feltkamp1,
- Siamaque Kazem1,
- Els van der Meijden1,
- Chris Lauber1and
- Alexander E. Gorbalenya1,2
+Author Affiliations
1. 1Department of Medical Microbiology, Leiden University Medical Center, Leiden, The Netherlands 2. 2Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, 119899 Moscow, Russia