 Although arthropods are important viral vectors, the biodiversity of arthropod viruses, as well as the role that arthropods have played in viral origins and evolution, is unclear. Through RNA sequencing of 70 arthropod species we discovered 112 novel viruses that appear to be ancestral to much of the documented genetic diversity of negative-sense RNA viruses, a number of which are also present as endogenous genomic copies. With this greatly enriched diversity we revealed that arthropods contain viruses that fall basal to major virus groups, including the vertebrate-specific arenaviruses, filoviruses, hantaviruses, influenza viruses, lyssaviruses, and paramyxoviruses. We similarly documented a remarkable diversity of genome structures in arthropod viruses, including a putative circular form, that sheds new light on the evolution of genome organization. Hence, arthropods are a major reservoir of viral genetic diversity and have likely been central to viral evolution.
Although arthropods are important viral vectors, the biodiversity of arthropod viruses, as well as the role that arthropods have played in viral origins and evolution, is unclear. Through RNA sequencing of 70 arthropod species we discovered 112 novel viruses that appear to be ancestral to much of the documented genetic diversity of negative-sense RNA viruses, a number of which are also present as endogenous genomic copies. With this greatly enriched diversity we revealed that arthropods contain viruses that fall basal to major virus groups, including the vertebrate-specific arenaviruses, filoviruses, hantaviruses, influenza viruses, lyssaviruses, and paramyxoviruses. We similarly documented a remarkable diversity of genome structures in arthropod viruses, including a putative circular form, that sheds new light on the evolution of genome organization. Hence, arthropods are a major reservoir of viral genetic diversity and have likely been central to viral evolution.
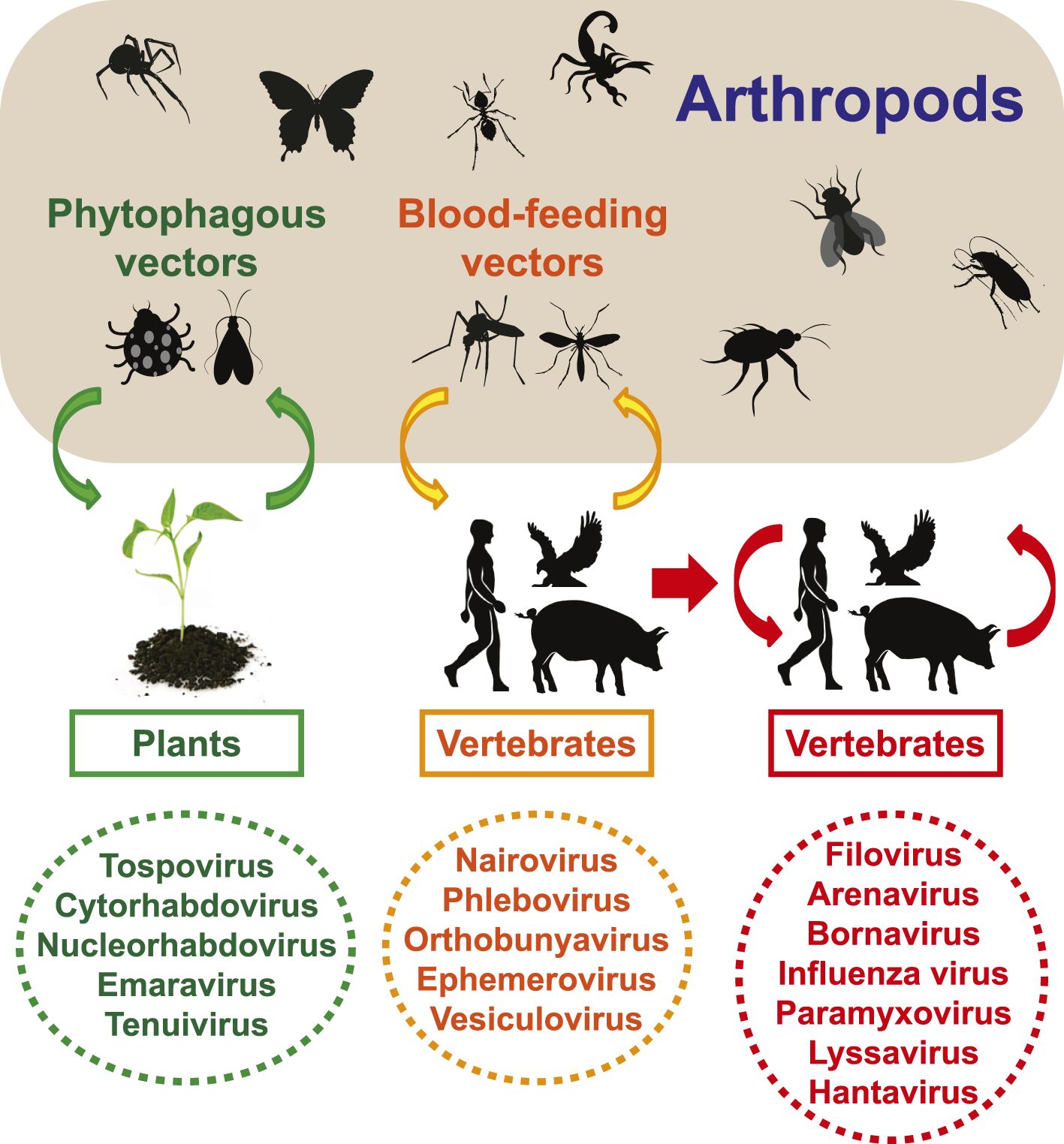
Negative-sense RNA viruses are important pathogens that cause a variety of diseases in humans including influenza, hemorrhagic fever, encephalitis, and rabies. Taxonomically, those negative-sense RNA viruses described to date comprise at least eight virus families and four unassigned genera or species (King et al., 2012). Although they share (i) a homologous RNA-dependent RNA polymerase (RdRp), (ii) inverted complementary genome ends, and (iii) an encapsidated negative-sense RNA genome, these viruses display substantial diversity in terms of virion morphology and genome organization (King et al., 2012). One key aspect of genome organization is the number of distinct segments, which is also central to virus classification. Among negative-sense RNA viruses, the number of segments varies from one (orderMononegavirales; unsegmented) to two (familyArenaviridae), three (Bunyaviridae), three-to-four (Ophioviridae), and six-to-eight (Orthomyxoviridae) and is further complicated by differences in the number, structure, and arrangement of the encoded genes.
Despite their diversity and importance in infectious disease, the origins and evolutionary history of the negative-sense RNA viruses are largely obscure. Arthropods harbor a diverse range of RNA viruses, which are often divergent from those that infect vertebrates (Marklewitz et al., 2011,2013;Cook et al., 2013;Ballinger et al., 2014;Qin et al., 2014;Tokarz et al., 2014a,2014b). However, those arthropod viruses sampled to date are generally those that have a relationship with vertebrates or are known to be agents of disease (Junglen and Drosten, 2013). To determine the extent of viral diversity harbored by arthropods, as well as their evolutionary history, we performed a systematic survey of negative-sense RNA viruses using RNA sequencing (RNA-seq) on a wide range of arthropods.
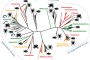
We focused our study of virus biodiversity and evolution on 70 potential host species from four arthropod classes: Insecta, Arachnida, Chilopoda, and Malacostraca (Table 1andFigure 1). From these samples, 16 separate cDNA libraries were constructed and sequenced, resulting in a total of 147.4 Gb of 100-base pair-end reads (Table 1). Blastx comparisons against protein sequences of negative-sense RNA virus revealed 108 distinct types of complete or nearly complete large (L) proteins (or polymerase protein 1 (PB1) in the case of orthomyxoviruses) that encode the relatively conserved RdRp (Tables 2–4). Four additional types of previously undescribed RdRp sequence (>1000 amino acids) were identified from the Transcriptome Shotgun Assembly (TSA) database. Together, these proteins exhibited an enormous diversity in terms of sequence variation and structure. Most notably, this data set of RdRp sequences is distinct from both previously described sequences and from each other, with the most divergent showing as little as 15.8% amino acid sequence identity to its closest relatives (Tables 2–4). Overall, these data provide evidence for at least 16 potentially new families and genera of negative-sense RNA viruses, defined as whose RdRp sequences shared less than 25% amino acid identity with existing taxa.
Full text is here:
http://elifesciences.org/content/4/e05378#ref-12