Аналіз вірому реципієнтів виявив новий вірус, який має спільні геномні властивості з Гепацівірусами (Hepaciviruses) і Пегівірусами (Pegiviruses)
Огляд
З метою дослідження трансмісії нових інфекційних агентів через кров, ми вивчили зміни віромного складу у реципієнтів до і після переливання крові. Завдяки цьому підходу ми змогли ідентифікувати і дати генетичну характеристику новому вірусу – hepegivirus 1 (HHpgV-1), який має спільні риси з вірусом гепатиту С та pegivirus (HPgV; раніше його називали вірус гепатиту G). Вірус гепатиту С (HCV) та пегівірус (HPgV) належать до роду Hepacivirus та Pegivirus родини Flaviviridae. Гепегівірус 1 типу (HHpgV-1) був виявлений в сироватці крові двох реципієнтів та двох пацієнтів з гемофілією, що отримали певну кількість фактору згортання крові. Раніше вірус ідентифікували тільки в зразках після переливання крові, і вважали, що він передається шляхом трансфузії. У хворих на гемофілію спостерігалась вірусемія протягом 201 і 1981 дня. 5′ не транслюючий регіон (UTR) гепегівірусу 1 типу містив IV тип ділянки внутрішньої посадки рибосоми (IRES), структурно схожий до послідовності вірусу гепатиту С та інших вірусів гепатиту. Однак філогенетичний аналіз не структурних генів (NS3 та NS5B) показав, що гепегівірус 1 типу формує монофілетично різну гілку від пегівірусів, а також його гомологи інфікують інший вид ссавців. Гепегівірус 1 типу (як і інші пегівіруси) уражує гризунів і рукокрилих. Геном гепегівірусу 1 типу кодує короткі високо основні білки лівого оперона E1, які виконують функцію кору в пакуванні РНК під час збірки віріону. Ідентифікація гепегівірусу 1 типу розширила знання про ряд геномних конфігурацій вірусів і в майбутньому може призвести до повторного перегляду початкових критеріїв визначення роду Hepacivirus і Pegivirus.
Важливо
Щорічно в США під час переливання крові передається більш ніж 30 млн. компонентів. Контроль інфекційних агентів в крові – основний ключ безпеки важливих ресурсів медицини і охорони здоров’я. В статті ми повідомляємо про ідентифікацію нового вірусу гепатиту С /GB (HCV/GB або GBV-подібний вірус), який було знайдено у зразках сироватки крові людей. Ми також виявили персистентну довгострокову віремію гепегівірусу 1 типу у двох пацієнтів хворих на гемофілію. Гепегівірус 1 типу унікальний тим, що має спільні генетичні характеристики як з високо патогенним вірусом гепатиту С, так і з непатогенним пегівірусом (GBV-C). Наші результати внесено до списку вірусів людини та запроваджено дані для розробки реагентів щодо вивчення трансмісії даного вірусу, попередження передачі вірусу та виникнення нових захворювань.
Переливання вторинних компонентів крові може зберегти життя та покращити стан здоров’я, але потребує певних заходів безпеки, щодо попередження передачі інфекційних агентів. Виділення контамінованих компонентів крові також потребує точного контролю за патогенними вірусами (1, 2). Своєчасне діагностування таких вірусів, як гепатиту С, В та ВІЛ в людей хворих на гемофілію та інших реципієнтів, може попередити появу симптомів захворювань з подальшим перебігом (3–8). Досягнення технологій секвенування сприяло розвитку ідентифікації нових вірусів (9) та запровадило широке діагностування вірусів людини і тварин (10–15). Генетична характеристика нових вірусів – це перший крок до їх молекулярної і біологічної характеристики в цілому. Секвенування нових вірусів не тільки допомагає оцінити біологічні властивості, але і дає можливість для розвитку молекулярних реагентів, які можуть бути застосовані для молекулярних, епідеміологічних та клінічних досліджень трансмісії вірусів та пов’язаних з ними захворюваннями (9).
Вірус гепатиту С уражує близько 200 млн. людей по всьому світу і являє собою генетично розмаїту групу РНК вірусів з подібними біологічними властивостями. Зазвичай вірус персистує в 20-30% випадків після проходження гострої інфекції (16, 17). Вірус гепатиту С – патоген глобального масштабу, оскільки існує велика кількість невиліковних прогресуючих хвороб, наприклад, важкі захворювання печінки, цироз та гепатокарцинома. Терапія вірусу базується на високовартісних антивірусних препаратах, які пригнічують реплікацію вірусу, саме тому попередження нового вірусного захворювання залежить від пріоритетів (18). Ідентифікація вірусу гепатиту С в 1989 році (19) стала ключовим моментом для розвитку щодо його діагностики та поширенню парентеральним шляхом. Інші генетично споріднені віруси до вірусу гепатиту С, в тому числі пегівірус (HPgV) та низка взаємопов’язаних вірусів, які уражують коней, гризунів, летючих мишей та мавп (розглянуто у 20 та 21). Пегівірус (HPgV) спричинює поширені хвороби людини (від 1 до 4% у здорових донорів) та персистує роками без будь яких проявів симптомів (22).
Hepacivirus (NPHV) було ідентифіковано у нижчих приматів та вперше у собак та коней, що виявило широке коло хазяїв гепацівірусної інфекції (hepacivirus infection) (23–25). Зовсім недавно було визначено низку хазяїв гепацівірусів (hepaciviruses), серед яких коні, летючі миші, гризуни та давні примати (11, 12, 26–28). Схожим чином було знайдено та ідентифіковано нові пегівіруси, наприклад пегівіус летючих мишей у 2010 році (29), згодом пегівіврусу було знайдено у коней та гризунів (11, 12, 26, 27, 30). Нові відкриття розширили наші знання про різноманіття та хазяїв гепацівірусів та пегівірусів (hepaciviruses and pegiviruses) (20). Однак, тільки два віруси уражують людину: гепатит С та пегівірус. Отже ми опишемо вперше ідентифікований людський гепегівірус 1 типу (HHpgV-1), який має спільні риси як з вірусом гепатиту С, так і з пегівірусом.
Результати
Аналіз вірому реципієнтів до і після трансфузії. Для того, щоб показати зміни віромного складу у реципієнтів, було досліджено ряд зразків до і після трансфузії, які були зареєстровані у відділі вивчення вірусів, що передаються шляхом трансфузії (Transfusion-Transmitted Viruses Study – TTVS) з липня 1974 року по червень 1980 року. Було взято зразки однієї серії 44 індивідів до та після переливання крові і проаналізовано склад нуклеїнових кислот, проведено ампліфікацію та секвенування (11). В цілому 154 серед 119 млн. зразків було вибрано серед до і після трансфузійних. Описова статистика свідчить про те, що стандартне та проміжне відхилення було розраховано для надлишку вірусів (viral abundance). Надлишок до і після переливання крові порівняли за критерієм суми рангів Уилкоксона. Порівняння двох зразків засвідчило про суттєве підвищення загальної кількості вірусів (P ‹ 0.009) та анеловірусів (anellovirus) (P ‹ 0.007) після переливання крові. (Fig. 1). Для того, щоб зрозуміти походження і склад анеловірусів, кількість яких зросла після переливання крові, було зроблено порівняльний аналіз всіх вірусів в зразках з шести TTVS до і після трансфузії та показано різне зростання кількості анеловірусів та шести зразків, в яких кількість анеловірусів зросла в 10 разів у післятрансфузійних зразках. Аналіз також показав, що більшість анеловірусів в зразках після переливання крові були іншого виду або генотипу, тобто генетично відрізнялись від тих, що були в зразках до переливання. Також було знайдено генетично споріднені зразки анеловірусів в до і після переливання крові (Fig. 1). Це може свідчити про те, що інфікування відбулося в обох випадах нестабільними анеловірусами, які також можуть перебувати у вигляді конфекцій. Серед всіх зразків приблизно 40% вірусів не було класифіковано через низьку подібність серед відомих (в діапазоні значень E value range, від 10-2 до 10-6). Подальший аналіз некласифікованих вірусів довів наявність нових флавівірусів в двох післятрансфузійних зразках.
Ідентифікація гепегівірусу 1 типу (HHpgV-1). Аналіз зразків двох сиквенсів, TTVS-772 та TTVS-790 (обидва взяті після переливання крові), підтвердив наявність нових флавівірусів, які в свою чергу подібні до вірусу гепатиту С та пегівірусу. ПЛР використано для підтвердження наявності у вихідній сироватці крові сиквенсів подібних до флавівірусів. Після проведення декількох аналізів зразків TTVS-772 та TTVS-790, які піддали більш ретельному секвенуванню та згруповали за >100 млн. кінцевих однойменних просвічувань. Всі проаналізовані послідовності подібні до геномів відомих гепацівірусів та пегівірусів, крім того, виявили подібність протеїнів (в діапазоні E value < 0.0001), різні фрагменти геномів даних вірусів об’єднали використовуючи ПЛР та розшифували первинну послідовність за допомогою методу Сангера. В результаті фрагмент геному, що кодує поліпротеїн гепегівірусу 1 типу, виявився в зразку TTVS-790. Також було синтезовано кДНК шляхом множинної ампліфікації (23) для того, щоб відтворити 9,538 нуклеотидів геному нового вірусу – гепегівірусу 1 типу.
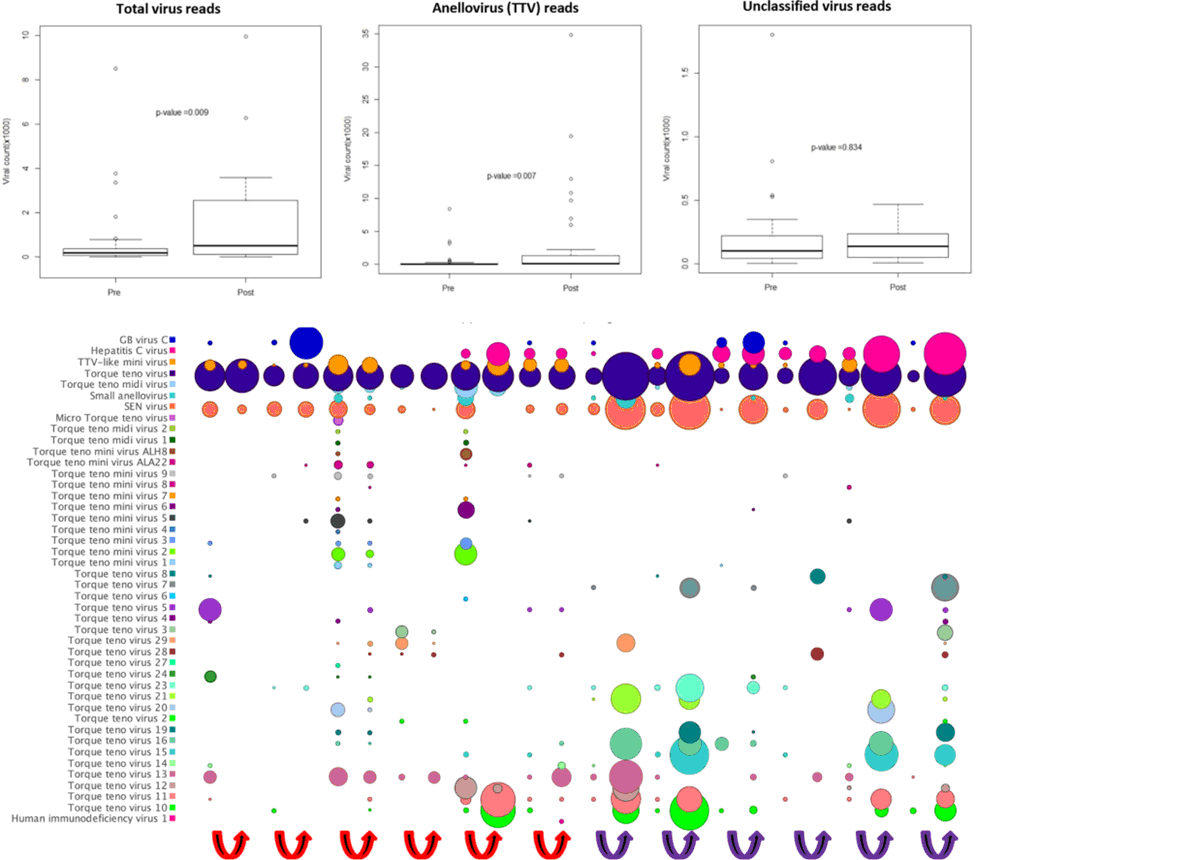
FIG.1 Аналіз вірому зразків TTVS до та після переливання крові. Зверху показано статистичний аналіз загальної кількості вірусних послідовностей (ліворуч), анеловірусів (посередині), і некласифікованих вірусних послідовностей (праворуч) в до- та посттрансфузійних зразках. Зверху також зображено метагеномний аналіз вірусних послідовностей в до- та посттрансфузійних зразках, відібраних серед 12 TTVS. Червоними стрілками позначено пари до- та посттрансфузійних зразків, які виявились однаковими; фіолетовими – різними.
Поліпротеїн кодуючий фрагмент геному гепегівірусу 1 типу. Геном гепегівірусу 1 типу містить безперервну відкриту рамку зчитування з кодоном термінації в позиції 9501 (Fig. 2). Передбачення стартового кодону поліпротеїну вірусу виявилось проблематичним, оскільки на 5′ кінці містилось декілька стартових триплетів АУГ (метіонін). Позиції 195-204 відрізняються від оптимальної послідовності Козак (ГТТЦ|АТГ|ГАГГ та АГГГЦ|АТГ|ЦЦЦА). Кодон в позиції 330 містив оптимальну послідовність (ГЦААЦ|АУГ|ГГГУ), четверту (позиція 519) – близька до оптимальної та п’яту (позиція 587), що не відповідає визначеній послідовності E1 пегівірусів. Визначення РНК послідовності 5′ нетрансльованого кінця дало змогу визначити те, що третій триплет АТГ (позиція 330) є ініціюючим і це було взято до уваги під час аналізу сайтів розщеплень білків.
Еволюційна спорідненість гепегівірусу 1 типу та гепацівірусів і пегівірусів. Відкрита рамка зчитування гепегівірусу 1 типу знаходиться в одній лінії на ряду з послідовностями гепацівірусів і пегівірусів взятих з людини та шимпанзе (HPgV, simian pegivirus infecting chimpanzees [SPgVtro], та HCV), нижчих приматів (SPgV-OWM та SHcV-OWM), пегівіруси GBV-A та GBV-B приматів нового світу (SPfV-NWM та SHcV-NWM), гризунів та різних варіантів мишей (RPgV та RHcV-1 to RHcV-3) та коней (EPgV-1, EPgV-2, та NPHV), декількох груп пегівірусів та гепацівірусів летючих мишей (BPgV-1 до BPgV-5 та BHcV-1 до BHcV-4). Відкрита рамка зчитування амінокислотної послідовності більшої частини геномів пегівірусів та неструктурних регіонів геному гепацівірусів розташовані в одну лінію. Проведено філогенетичний аналіз щодо встановлення еволюційного зв’язку гепегівірусу 1 типу з іншими групами на основі високо консервативних NS3- та NS5B-кодуючих регіонів. Побудовано два філогенетичних дерева на основі порівняння двох високо консервативних регіонів, де послідовність гепегівірусу 1 типу чітко вирізняється серед послідовностей гепацівірусів і пегівірусів (Fig.3). Обидва регіони розділились на дві окремі монофілетичні гілки згідно з класифікацією: на гепацівіруси і пегівіруси. Дві окремі монофілетичні гілки також було розділено на окремі гілки, що утворили дві групи (groups 1 та 2); група 1 містить варіанти хазяїв, такі як люди, примати, летючі миші та коні, диференційованих на лінії 1, 4 та 5, тоді як послідовність гепегівірусу 1 типу і варіанти хазяїв – гризуни, летючі миші розділені на 2 і 3 лінію та віднесені до групи 2.
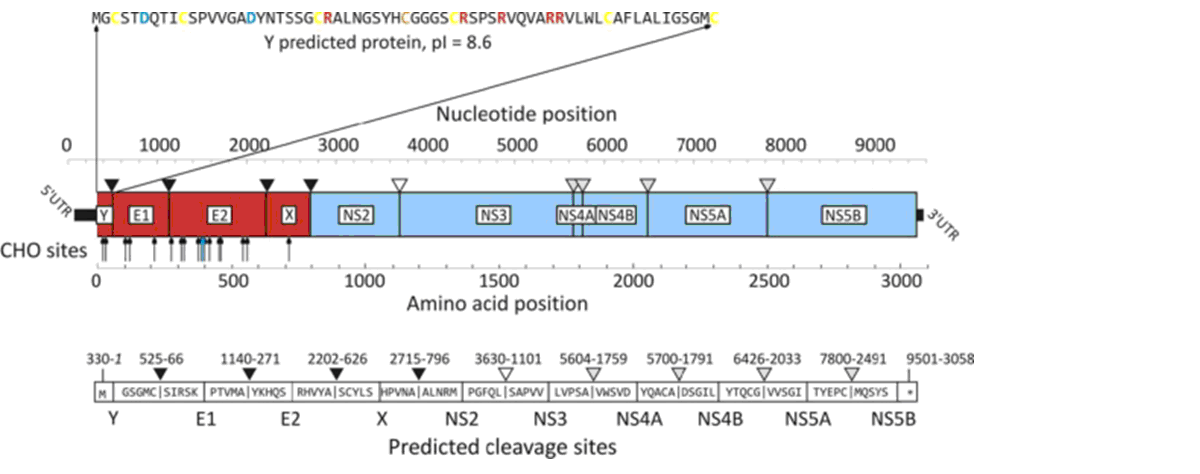
FIG.2 Організація геному та сайтів розщеплення протеїнів HHpgV-1. Позиції сайтів розщеплень NS2 (позначено білими трикутниками) та протеази NS3 (позначено сірими трикутниками), в геномах яких було передбачено гомологічні сайти неструктурних генів попередньо розглянутих пегівірусів і гепацівірусів. Сайти розщеплень в структурних регіонах геному (позначено чорними трикутниками) було незалежно передбачено, використовуючи сервер SignalP 4.1. Передбачені N-глікозильовані сайти протеїнів капсиду позначено білими стрілками; одиничні О-глікозильовані сайти позначено синіми стрілками. Амінокислотні послідовності Y протеїну показано над діаграмою геному. Заряджені амінокислоти та цистеїн позначено кольором.
Передбачення сайтів розщеплення білків гепегівірусу 1 типу. Крайні позиції геному гепегівірусу 1 типу було передбачено завдяки застосуванню декількох підходів. Відкрита рамка зчитування амінокислотної послідовності на 5՛ кінці геному піддали дії сигналу P 4.0 для ідентифікації сайтів розщеплень, що також використовувалось для ідентифікації структурних генів інших пегівірусів (Fig. 2). Це дало змогу виявити три сайти розщеплення білків в позиціях, які було до цього ідентифіковано для пегівірусу коней та приматів (SPgV-NWM). Однак, на додаток до E1, E2 та X протеїну, гепегівірус 1 типу повинен був ще містити 65 амінокислот та білкову цистеїн-збагачену ділянку вище E1. Також було ідентифіковано кодуючи ділянки у пегівірусів гризунів (12) та позначено “VR” в групі 2 – летючі миші (лінії 2 та 3) (26). Y або VR протеїни досить сильно варіювали в розмірах та не мали спільних послідовностей відносно одне одного. Послідовність гепегівірусу 1 типу все ж таки розділяла спільні властивості з декількома позитивно зарядженими рівнями осаду (ізоелектрична точка – pI 8.6) (Fig. 2), порівняно з попередньо проаналізованими осадами (pI від 8.2 до 9.1) та RPgV (pI 10.1). Y та VR протеїни відрізнялись в передбачених N-кінцевих ділянках, в тому числі Y/VR протеїни гризуна та летючої миші, які могли б транспортувати протеїни крізь ендоплазматичний ретикулум. Та навпаки, Y протеїн гепегівірусу 1 типу не мав специфічної транслокаційної послідовності, тобто протеїн локалізований в цитоплазмі. Схожим чином відбувається процесинг протеїнів кору у гепацівірусів, саме протеїни кору приймають участь у пакуванні РНК та формуванні віріонів. Зазначимо, що Y протеїн гепегівірусу 1 типу може виконувати схожу функцію.
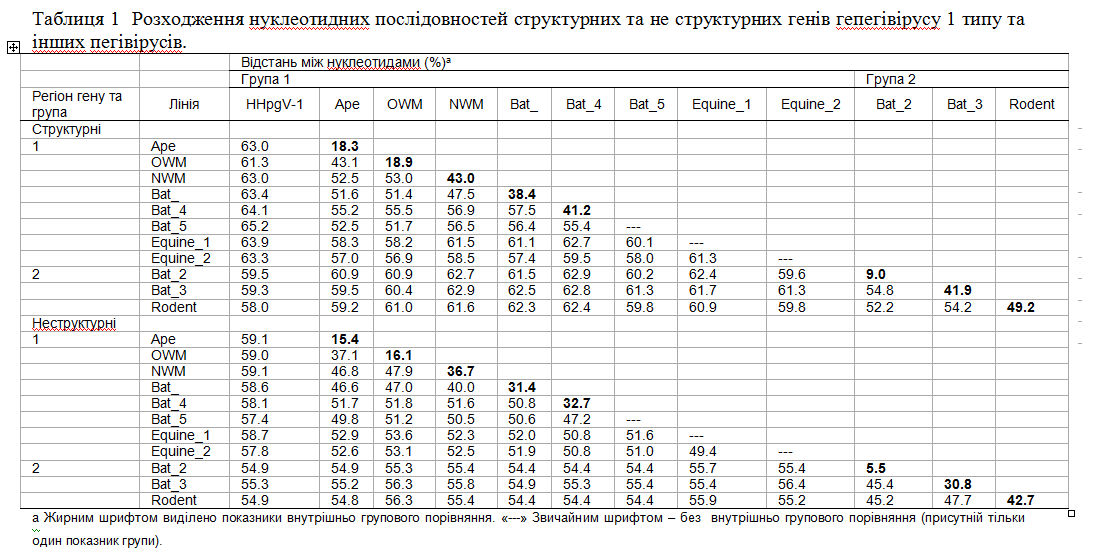
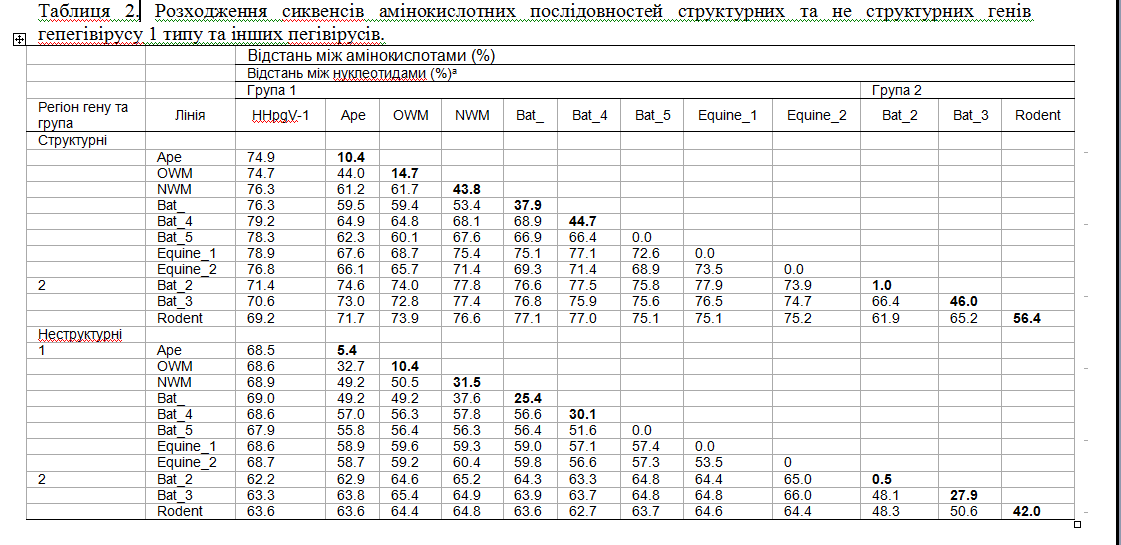
Протеїн E2 містив 11 взаємопов’язаних глікозильованих N-сайтів − така кількість перевищила фіксовані норми для інших пегівірусів; було ідентифіковано додаткові сайти у протеїну E1 (n = 3) та X (n = 1). E2 додатково містив O-сайт в позиції 422 (Fig.2). Частину глікозильованого сайту, в порівнянні з вірусом гепатиту С та деякими іншими гепацівірусами, було легко відрізнити від рідко фіксованих сайтів у людини та інших пегівірусів 1 групи. Для передбачення сайтів розщеплень серед неструктурних протеїнів було знайдено відповідні послідовності між гепегівірусом 1 типу та іншими пегівірусами з визначеними позиціями сайтів для ідентифікації (Fig.2). Протеїни, які було передбачено в фрагменті NS гепегівірусу 1 типу було взято для порівняння таких самих фрагментів у інших пегівірусів і гепацівірусів (Fig.2). Розходження між сиквенсами гепегівірусу 1 типу та іншими пегівірусами одного ряду було високим, від 61 до 65 %, а в групі 1, серед сиквенсів пегівірусів, розходження перебувало в межах від 58 до 60 % враховуючи структурний регіон гену групи 2 (Table1). Була встановлена відповідність між амінокислотними послідовностями в даній ділянці геному від 68 до 79% та від 69 до 71%. Неструктурні частини гену виявились більш консервативними, нуклеотидні відстані варіювали в межах від 57 до 59 % у групи 1 і 2 та між гепегівірусом 1 типу та групою 1 і 2 в межах 55 % , у взятих до уваги двох груп – від 68 до 69 % та від 62 до 64 % (Table 2). Зважуючи результати можна сказати, що розходження відбулось серед всіх послідовностей геному пегівірусу, та більше ніж 40 % припадає на багато віконний фрагмент гепегівірусу 1 типу та кожну лінію пегівірусів (Fig.4). Високий рівень дивергенції спостерігався в структурних регіонах гену та NS4B/NS5A, коли мінімальний рівень було знайдено в функціонально консервативних полімеразних і геліказних мотивах NS5B та NS3.
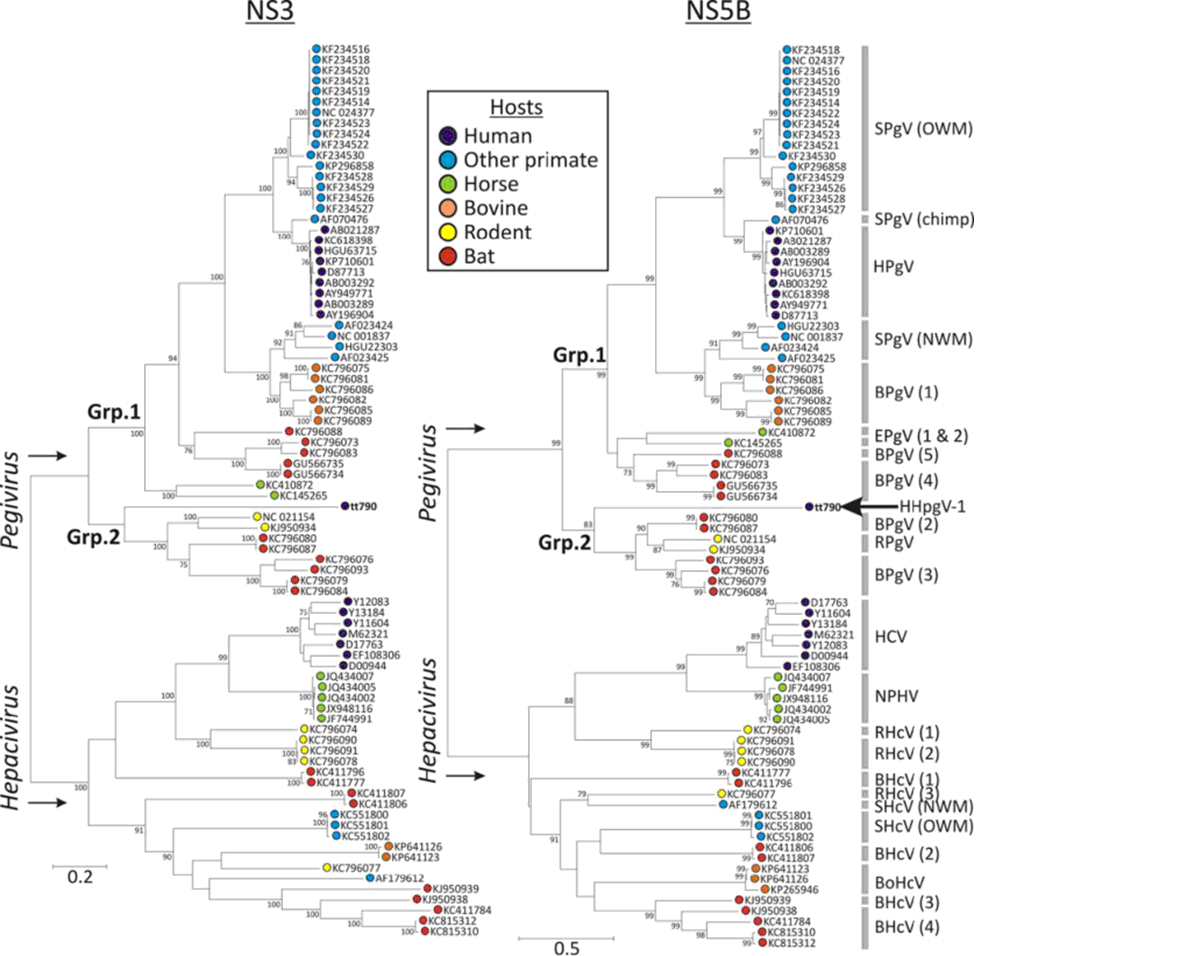
FIG. 3 Зображено максимально точний філогенетичний аналіз генів NS3 та NS5B гепегівірусу 1 типу (HHpgV-1) (tt790) та повні геномні послідовності інших гепацівірусів та пегівірусів, що в свою чергу уражують різні види ссавців. Філогенетичний аналіз для кожного об’єму інформації було зроблено, використовуючи формування вибірки методом бутстрепа для визначення правильності групування; фактичне значення показано на гілках. Абревіатура: гепацівірус (HcV); пегівірус (PgV); мавпи (OWM – Old World monkey); капуциноподібні мавпи (NWM – New World monkey); гризуни (R); летюча миша (B); жуйна тварина (Bo); гепацівірус неприматів (NPHV). Клітинні лінії показано в дужках.
Передбачення 5′ нетранслюючого регіону(UTR) структури РНК. Пегівіруси було розподілено на основі кодуючих фрагментів, в тому числі за спорідненістю 5′ UTR на дві групи окремих філогенетичних гілок. Всі послідовності групи 1 могли бути передбачені на основі вторинної структури РНК, яка в свою чергу близька до GBV-A та вже відомої структури пегівірусу коней (11). Несподівано, але ділянки 5′ UTR групи 2 пегівірусів не були гомологічними групі 1, однак мали певні кореляції. Незважаючи на те, що спостерігалось розходження між всіма іншими вірусними 5′ UTR послідовностями, деякі мотиви були спільними для гепацівірусів, в тому числі високо консервативна послідовність ТАЦАГЦЦТГАТАГГГТ в позиції 274. Послідовність лежить в основі IIIe домену (регіон у вигляді псевдопетлі) гепацівірусів та належить до ділянки внутрішньої частини рибосоми (internal ribosome entry site – IRES). Порівнюючи 5′ UTR гепегівірусу 1 типу та гепацівірусів, аналіз показав наявність стандартної IRES 4 типу (Fig.5) та позицією вище від домену 1 і 2 вірусу гепатиту С, – ‘петлю на стеблі’. Однак miR-122, в основі якої лежить послідовність АЦУЦУЦЦА, була відсутня у гепегівірусу 1 типу та інших пегівірусів. Як було передбачено, інша група 2 пегівірусів мала подібну структуру до типу 4 IRES елементів, не дивлячись на їх стандартне розходження (дані не показано). В даній структурній моделі РНК гепегівірусу 1 типу, третій триплет АТГ в позиції 330 (як і в оптимальній послідовності Козак) нижче від псевдопетлі в домені IV розташовано 11 нуклеотидів, та за аналогією це імовірний кодон початку ініціації гепегівірусу 1 типу.
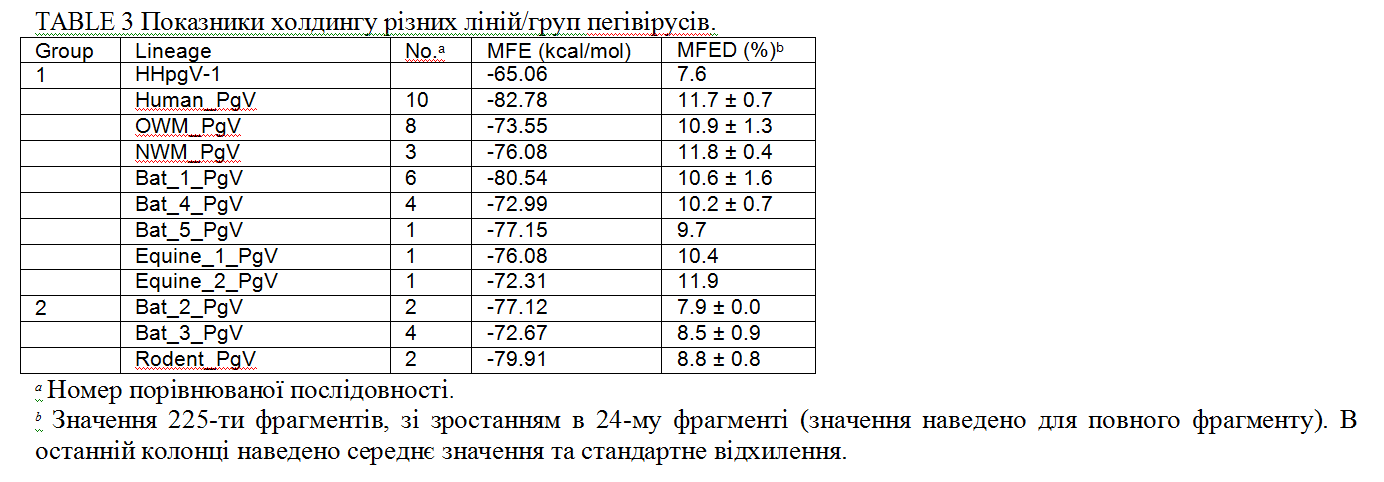
GORS. Термодинамічний аналіз фолдингу послідовності гепегівірусу 1 типу показав зміну вільної енергії на 7.6% між мінімальною енергією фолдингу та невпорядковано розташованою послідовністю (MFED); дослідження було узгоджено з наявністю повногеномної впорядкованої структури РНК (genome-scale ordered RNA structure – GORS) гепегівірусу 1 типу (23). Однак, мінімальна вільна енергія невпорядковано розташованої послідовності (MFED) була нижчою ніж у представників групи 1: пегівірусу (середнє значення, 11.7%; стандартне відхилення, 0.7%), SPgV-OWM (10.9% ± 1.3%), SPgV-NWP (11.8% ± 0.4%), EPgV-1 (10.4%), та EPgV-2 (11.9%). Середнє значення MFED гепегівірусу 1 типу порівняли з середнім значенням MFED пегівірусів групи 2 (від 7.9% до 8.8%) (Tаблиця 3).
Інфекції, які викликає гепегівірус 1 типу, їх запобігання та конфекції. Для того, щоб визначити природу інфекцій, які викликає гепегівірус 1 типу в людей, було проаналізовано зразки до та після переливання крові з 46 по 116 TTVS. Також було проаналізовано зразки з центру контролю захворювань пацієнтів з гемофілією (Multicenter Hemophilia Cohort Study – MHCS). Тільки два посттрансфузійні зразки взяті з TTVS були позитивним на РНК гепегівірусу 1 типу : TTVS-772 та TTVS-790. РНК позитивний зразок TTVS-772 було взято у пацієнта після 17 днів трансфузії. Зразок TTVS-0927 було взято у того ж пацієнта за один день до переливання крові та інший зразок TTVS-1095 було взято після 281 дня трансфузії, який виявився негативним на РНК гепегівірусу 1 типу. Зразок TTVS-790 було взято після 7 днів трансфузії. Зразок TTVS-1608 було взято в того ж самого пацієнта за день до переливання крові та зразок TTVS-791 було взято після 241 дня трансфузії, який в свою чергу виявився негативним на РНК гепегівірусу 1 типу. Отримані результати свідчать про вірусемію гепегівірусу 1 типу в зразках після переливання крові, з подальшим кліренсом вірусу через імунну відповідь в обох інфікованих пацієнтів.
З 106 відібраних зразків лише два виявились РНК позитивними на гепегівірус 1 типу – M3127 та M4287. Також було взято два допоміжні зразки, які було взято з різницею в 201 день, один зразок було відібрано за 144 дня перед тим, як відібрати позитивний зразок та два інші зразки на 560 і 3461 день після того, як було відібрано другий негативний зразок. Другий зразок позитивний на гепегівірус 1 типу, тобто M4287, до якого було відібрано чотири позитивні зразки. Три зразки з чотирьох було відібрано на 399, 742 та 1981 дні після першого дня відбору. Ці результати свідчать про вірусемію, яка триває більше ніж 201 та 1981 (5.4 роки) день.
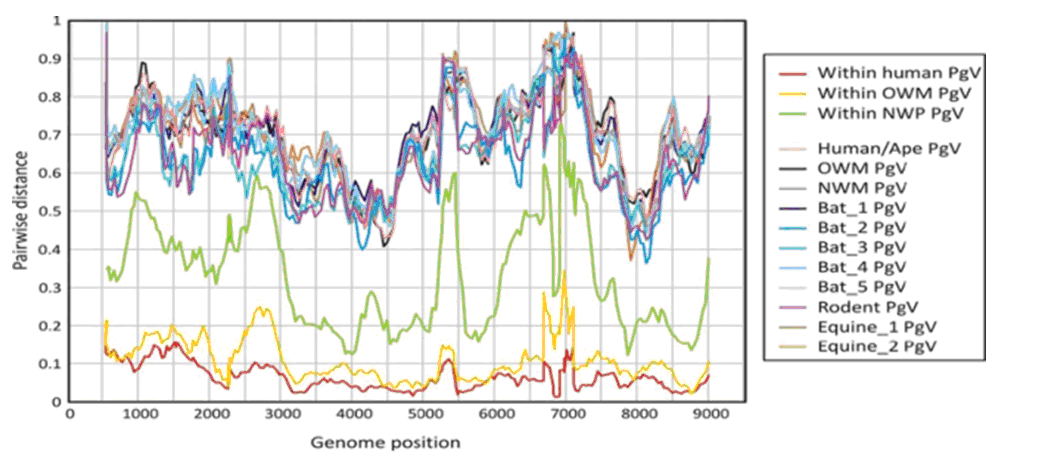
FIG. 4 Розходження амінокислотних послідовностей гепегівірусу 1 типу та інших пегівірусів. Діаграму було зроблено на основі 240 нуклеотидної послідовності з 24 нуклеотидною послідовністю, що поступово зростає (проміжну точку позначено на осі х). Для проведення порівняння було також взято відстані між пегівірусами мавпи і людини. Розходження послідовності гепегівірусу було позначено номером AF121950. OWM; NWP; NWM.
Всі зразки було секвеновано в обох напрямках, для того, щоб підтвердити результати та встановити генетичну різноманітність між різними варіантами гепегівірусів 1 типу. Філогенетичний аналіз підтвердив близьку генетичну спорідненість обох варіантів гепегівірусу 1 типу: MHCS та TTVS зразків (Fig. 6). Однак, обидва варіанти в порівнянні з пегівірусами та гепацівірусами мають генетичну варіабельність. Коінфекції, викликані HCV та HPgV було ідентифіковано завдяки скринінгу зразків позитивних на HHpgV-1. Три зразки гепегівірус позитивні з чотирьох мали конфекцію з HCV (з генотипом 1а в двох зразках та генотипом 1b в одному). Всі зразки з гепегівірусом 1 типу та їх відповідні зразки не мали пегівірус негативну коінфекцію.
Обговорення результатів
Було використано об’єктивний підхід для дослідження вірусів та їх характеристики, для того, щоб показати ризик трансфузії (9). Також було досліджено ряд зразків до та після трансфузії з TTVS та проведено порівняльний аналіз для виявлення змін віромів. Порівняльний аналіз показав, що кількість анеловірусів (anelloviruses) зросла після трансфузії (Fig.1). Зразки TTVS було досліджено в реципієнтів раніше (31, 32), але структурний аналіз та повне секвенування, які застосовувались раніше, не підходили для характеристики скла зразків TTVS протягом всього часу.
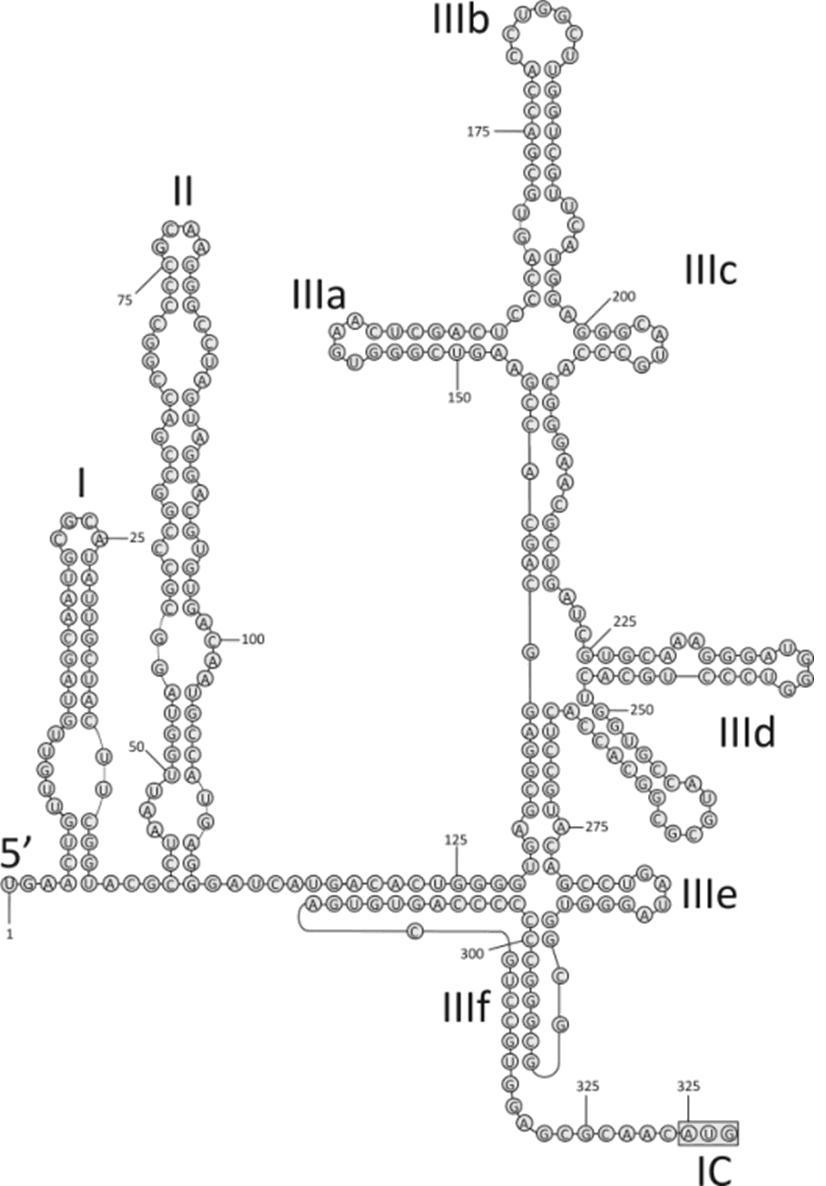
FIG 5 Вторинну структуру РНК з 5′ UTR було передбачено на основі визначення знаходження генів гепегівірусу 1 типу та гепацівірусів залежно від петельного домену IIIe. Петельні домени I та II було передбачено, використовуючи Mfold.
Може бути декілька причин зміни складу вірому, які спостерігались, що пов’язані зі зниженням загального стану здоров’я реципієнтів та появою нових інфекцій всередині лікарні, включаючи передачу вірусів під час переливання крові. Найцікавішим дослідженням виявилась наявність у зразках після переливання крові нового вірусу людини – гепегевірусу 1 типу (HHpgV-1). Доступність зразків сироваток крові з TTVS дала можливість з’ясувати природу інфекції, яку викликає гепегівірус 1 типу; вірусна інфекція була присутня тільки в посттрансфузійних зразках та згодом зникала через імунну відповідь організму. Не дивлячись на те, що отримані результати свідчать про можливість передачі гепегівірусу 1 типу шляхом трансфузії, потрібно розвивати подальші дослідження щодо пегівірусу 1 типу, його наявності в посттрансфузійних зразках та можливості викликати ятрогенні інфекції. Якщо присутність нового вірусного геному у сироватці крові людини підтверджує інфекцію, все одно потрібно шукати нові дані, включаючи показники конверсії сироватки крові інфікованих пацієнтів; саме тому ми розвиваємо серологічний аналіз гепегівірусу 1 типу (23, 33). Ми також приділяли значну увагу аналізу клінічних зразків в лабораторії, так як контаміновані зразки з нуклеїновими кислотами можуть слугувати основним ресурсом дивергенції вірусів (9, 34). Обов’язковим було включення контрольних негативних зразків під час проведення досліджень. Контрольні зразки, які було досліджено, екстрагували водою та фосфатно-сольовим буфером; теж саме було пророблено з контрольними реагентами для зворотної транскрипції та ПЛР скринінгу. Більш того, порівняння різних ампліфікованих зразків гепегівірусу 1 типу, що були відібрані в різних пацієнтів у різний час дозволило виявити певне розходження, в основному сконцентроване в схожих сайтах геному; було виявлено специфічні послідовності, за якими зразки розподілили на відповідні групи (Fig.6). Гепегівірус 1 типу було виявлено в посттрансфузійних зразках в двох з 46 TTVS за допомогою секвенування. ПЛР скринінг посттрансфузійних зразків взятих 70 TTVS не дозволив виявити подальші позитивні. Гепегівірус 1 типу було виявлено тільки у пацієнтів, хворих на гемофілію (2 з 106 зразків). HHpgV-1 виявлявся доволі рідко, в порівнянні з HCV та HPgV в групах риску. Це можна пояснити декількома шляхами: 1) інфекція, яку викликає гепегівірус 1 типу не настільки поширена, 2) сам по собі вірус персистує доволі рідко; 3) ПЛР скринінг, що було використано для виявлення вірусемії не дав змоги ампліфікувати послідовності, що мали більше генетичне розходження в порівнянні з пегівірусом 1 типу (35). Останні дослідження призвели до недооціненої можливості попередити інфекцію, що викликає вірус гепатиту С, і все завдяки ідентифікації різних генотипів цього вірусу. Контроль частоти виникнення інфекцій, що викликає гепегівірус 1 типу та їх попередження потребує додаткових молекулярних та серологічних досліджень.
Організація геному HHpgV-1 дозволила виявити нові генетичні аспекти в цьому вірусі людини. По перше, ділянка 5′ UTR не гомологічна іншим як таким у пегівірусів, також вона сформувала IRES 4-го типу, що фактично була описана лише для Hepacivirus та Pestivirus родини Flaviviridae. Гепегівірус 1 типу мав спільні послідовності з двома іншими групами пегівірусів, не дивлячись на їх розходження між собою (12, 26). Це відкриття доповнює раніше описану послідовність IRES, подібну до пегівірусів та знайдену в гепацівірусів летючих мишей (27). Так само з пікорнавірусами (36), відбувався обмін між функціональними частинами трансляції далеких родичів і очевидно, що цей процес відбувається часто в еволюції РНК вмісних вірусів, незважаючи на рушійні сили та вплив різних вірусних послідовністей IRES на реплікацію та механізм проникнення в клітину хазяїна, які в більшості випадків залишаються невідомими. Однак, існує певна залежність між різними типами IRES та факторами ініціації трансляції, таких як eIF3 та eIF-2α (37, 38), а це в свою чергу впливає на здатність протистояти вимкненню процесу трансляції в клітині хазяїна за допомогою стресової відповіді та індукцією інтерферон-стимулюючих факторів (39). Іншою особливістю геному гепегівірусу 1 типу є присутність короткої кодуючої послідовності вище від E1 (Fig.3). Цей регіон кодує цитоплазматичний протеїн, коротший за поверхневий білок вірусу гепатиту С, який може приймати участь в упаковці РНК та збірці віріонів. Гепегівірус 1 типу подібний до вірусу гепатиту С та інших гепацівірусів в положенні певних N-глікозильованих сайтів, а також в передбачуванні генів капсиду, а також послідовності E2, в якій присутні 11 N-сайтів та один передбачений O-сайт. Ця надмірно глікозильована частина контрастує з недостатністю таких сайтів у гепегівірусів та більшості інших пегівірусів, які слугують для них захистом від антитіл (40, 41), але вони допомагають вірусу проникати в гепатоцити (42). Припускають, що гепегівіруси 1 типу мають однакові механізми проникнення.
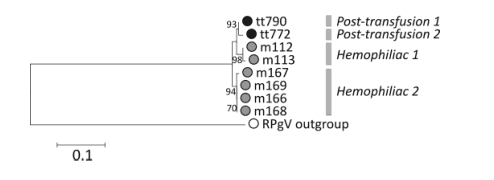
FIG 6 Філогенетичний аналіз послідовності NS3 гепегівірусу 1 типу різних зразків, відібраних з різних сироваток крові людини з використанням пегівірусу гризунів як вихідну групу. Філогенетичний аналіз зроблено, використовуючи формування вибірки методом бутстрепа для визначення правильності групування. Варіанти гепегівірусу 1 типу, які було знайдено в сироватках крові одних і тих же пацієнтів, позначено з правого боку (tt, TTVS; m, MHCS).
Вірусемія з персистенцією була виявлена у хворих на гемофілію у зразках, що були відібрані з певним інтервалом у часі з аглютинацією цих інфекційних зразків в концентраціях (від 1989 до 1990), що свідчить про схильність гепегівірусу 1 типу до довгострокової персистенції, до чого також схильна більшість гепацівірусів та пегівірусів. Гепегівірус 1 типу схожий з іншими вірусами цього роду РНК структурою розглянутою раніше – GORS, яка тісно асоційована з персистенцією +РНК вмісних вірусів (43, 44) різних родин ссавців. Не дивлячись на те, що ця спорідненість слабко зазначена (45), гепегівірус 1 типу маає спільні проміжні невпорядковано розташовані послідовності (MFED) з групою 2 пегіврусів та вірусу гепатиту С (Table 3). Подальша біоінформатична характеристика структурних конфігурацій вірусних РНК та особливо експериментально виділеного RPgV, допоможуть з’ясувати генетичні властивості персистуючих РНК вмісних вірусів.
Нещодавнє виявлення великої різноманітності сиквенсів гепацівірусів і пегівірусів та існування мозаїчності та варіабельності ознак певних геномів вірусів може призвести до повторної перевірки критерій походження родів гепацівірусів і пегівірусів (22). Як вже розлядалось вище, положення типу 4 IRES не визначає гепацівіруси, так як цей елемент присутній у всієї групи 2 пегівірусів (Fig. 4). Схожим чином деякі гепацівіруси мають подібний до пегівірусу 5′ UTR сиквенс. Наявність кор протеїну могла б бути визначальною ознакою наряду з пегівірусами, які мають кодуючу ділянку вище E1 та, у випадку гепегівірусу 1 типу – потенціально визначену цитоплазматичну локалізацію аналогічну до протеїну кору гепацівірусів. Незважаючи на все це, філогенетичний аналіз консервативних генів в неструктурних регіонах геному має чіткий розподіл, тобто є самостійні групи пегівірусів і гепацівірусів, в той час як структурні гени E1/E2/X пегівірусів малоподібні або взагалі не подібні до генів кодуючих протеїни E1 та E2. Подальша характеристика вірусів широкого кола хазяїв дозволить отримати більше інформації щодо цінності та довговічності сучасних критеріїв характеристики родів Hepacivirus and Pegivirus.
Повний аналіз вірому допоміг виявити новий вірус людини – гепегівірус 1 типу. Також слід відмітити, що 40% послідовностей всього проаналізованого вірому виявились некласифікованими через низьку спорідненість до вже відомих послідовностей вірусів. Віруси виявили помітну різноманітність за часом реплікації, експресії та генетичною складністю (46). Після отримання генетичної послідовності гепегівірусу 1 типу, було відображено некласифіковані послідовності та відмічено те, що декілька з них були частиною геному гепегівірусу 1 типу (кор, E1, E2, NS4A та NS4B гени). Ці відкриття свідчать про те, що інша частина некласифікованих послідовностей може являти собою невідомі віруси або ж їх геноми. База даних геному гепегівірусу 1 типу допоможе спрогнозувати молекулярні реактиви для вивчення попередження можливих захворювань, персистенції, їх передачі та вірус асоційованих хвороб людини. Крім того, запровадження інформації щодо нових вірусних захворювань людини, в тому числі ідентифікацію гепегівірусу 1 типу, розширює знання про походження, генетичну різноманітність та еволюцію гепаці- та пегівірусів.
Матеріали та методи
Зразки, відібрані у людей. Всі досліджені зразки було отримано з Національного центру дослідження серця, легень і крові та в свою чергу поділено на дві частини: TTVS та MHCS. Розроблений план дослідження було схвалено медичним центром при Колумбійському університеті (no. IRBAAAN5157). Зразки TTVS було відібрано в чотирьох центрах донорства США протягом липня 1974 року та червня 1980 року. MHCS-I також містили зразки хворих на гемофілію або ті, що мають певні порушення факторів згортання крові (з 1982 по 1986). Сироватки крові дорослих та дітей, що мали вроджені вади факторів згортання крові (гемофілія А або В – порушення факторів згортання крові VIII або IX), хворобу Вілебранда-Юнгенса або інші хвороби, було взято з восьми центрів захворювань на гемофілію в США в період між 1982 та 1985 рр. В дослідженні додатково прийняли участь 4 центри США та 4 європейські центри в період між 1987 та 1990 рр. Зразки MHCS-I було відібрано один раз кожного півроку з подальшим медичним аналізом, висновками та флеботомією (4–8).
Секвенування, ампліфікація та біоінформатичний аналіз вірому. Сироватки крові було профільтровано (фільтр діаметром 0.45-μm) та оброблено нуклеазами для звільнення від вірусних нуклеїнових кислот, потім екстраговано буфером NucliSens з використанням автоматизованої easyMAG системи (bio-Mérieux, United States). Тотальну РНК було транскрибовано зворотньою транскриптазою за допомогою SuperScript III kit (Invitrogen Life Technologies – технологія In vitro), використовуючи різні гескамерні праймерні. РНКаза Н виступила в ролі кДНК, другий ланцюг ДНК було синтезовано використовуючи фрагмент Кленова (3′ – 5′ негативні екзонуклеази) (New England Biolabs). Дволанцюгову ДНК було розділено на фрагменти довжиною 200-bp в середньому за допомогою ультразвукового приладу Covaris E210. Розділену ДНК було очищено та використано для конструкції в бібліотеці Illumina з використанням мови програмування Капа (KK8234; Kapa Biosystems) та SeqCap EZ бібліотеки SR (Nimblegen, Roche). Кількісну оцінку секвенованих зразків дала змогу встановити програма Agilent Bioanalyzer 2100. Зразки з низькими концентраціями було ампліфіковано шляхом збільшення кількості циклів ПЛР від 9 до 14. Секвенування було проведено на базі Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) з процесивністю в середньому 150 млн. зчитувань на стандартний фрагмент. Аналіз вірому включав тільки ті послідовності, які були найбільш споріднені до вірусів хребетних, саме тому було використано MEGAN version 5.10.5. Дані послідовностей було розподілено на окремі файли за допомогою програмного забезпечення Illumina та згруповано в файли FastQ для індивідуальних зразків. Послідовності було профільтровано використовуючи Q30 та локалізовано відповідно до геномів з GenBank за допомогою Bowtie2 mapper 2.0.6 (http://bowtie-bio.sourceforge.net). SAMtools (v 0.1.19) використано для згрупування консенсусних послідовностей геномів та статистики перекриття генів. Integrative Genomics Viewer (v 2/3/55; Broad Institute) використано для згрупування ділянок перекриття генів. Отримані послідовності було ідентифіковано, використовуючи карти секвенування Bowtie2 на противагу рекомендованих геномів бази даних NCBI. Дані, отримані шляхом секвенування зразків, було попередньо оброблено, використовуючи програмне забезпечення PRINSEQ (v 0.20.2). Виділені послідовності в подальшому зібрано de novo за допомогою MIRA (v 4.0) або SOAPdenovo2 (v 2.04), потім унікальні послідовності без повтору виділено для пошуку гомологічних, використовуючи базу даних MegaBlast на противагу GenBank. Послідовності, які виявили низьку гомологію або були не гомологічні заданим, аналізували за допомогою BLASTx на противагу GenBank (база білків). Вірусні послідовності, проаналізовані в BLASTx було проаналізовано в GenBank, для того, щоб виправити відхилення значення Е щодо меншого значення, в результаті чого було переглянуто базу даних та таксономію. Базуючись на ідентифікованих контигах для різних вірусних послідовностей, було завантажено послідовності з GenBank та використано їх для побудови генетичних карт, включаючи ділянки перекриття та повні геноми. Стандартне відхилення та середнє значення, згідно дискретної статистики, було розраховано для кількості вірусних частинок та їх характеристики. Співвідношення між зразками сироваток крові до та післі переливання порівняли за критерієм суми рангів Уїлкоксона.
Скринінг геному HHpgV-1
Поліпротеїн та нетранслюючу ділянку (UTR) гепегівірусу 1 типу було поставлено в один ряд з відомими гепаці- та пегівірусами. Нуклеотидні та амінокислотні мотиви виявили гомологію серед різних вірусних ліній, які потім було використано для пошуку праймерів щодо подальшого скринінгу зразків гепегівірусу 1 типу та пов’язаних варіантів. Для всіх сумішей ПЛР використано AmpliTaq Gold 360 master mix (catalog no. 4398881; Applied Biosystems) та 2 μl кДНК. Для ПЛР гепегівірусу 1 типу використовували напівгніздові праймери, в першому колі використали два праймери HHpgV-ak1 (5′-GTTGTATTCGCCACAGCCAC-3′) та HHpgV-ak2 (5′-TCAAAGTTTCCTGTGTAGCCTGT-3′), а в другому колі – HHpgV-ak3 (5′-GTATTCGCCACAGCCACTCC-3′) та HHpgV-ak2. Денатурація під час першого кола ПЛР відбулась протягом 8 хв. за t 95°C, надалі 10 циклів за t 95°C по 40 сек, 1 хв. за 56°C, 1 хв. за 72°C, 30 циклів по 30 сек. за 95°C, 1 хв. за 55°C та 1 хв. за 72°C і на фінальній стадії 5 хв. за 72°C. Перші 10 циклів температуру знижували на 0.5°C на кожний цикл, для того, щоб збільшити резистентність праймерів до мутацій протягом гібридизації. Другий цикл ПЛР було виконано за t 95°C протягом 8 хв., 10 циклів за t 95°C кожні 40 сек., 60°C протягом 1 хв. та за 72°C протягом 1 хв., 30 циклів за 95°C кожні 30 сек., за 56°C протягом 1 хв. та за 72°C протягом 1хв., на фінальній стадії за 72°C протягом 5 хв. Для проведення ПЛР вірусу гепатиту С було використано в першому циклі два праймери HCV-ak-F1 (5′-GCGCCCATCACGGCITAYGC-3′) та HCV-ak-R1 (5′-GTCTTGGTCCACRTTGGTRTACAT-3′), а в другому циклі – HCV-ak-F2 (5′-GCCCATCACGGCGTAYGCNCARCA-3′) та HCV-ak-R2 (5′-CTTGGTCCACRTTGGTRTACATYTG-3′). Перший цикл тривав 8 хв. денатурації за 95°C, подальші 10 циклів за 95°C кожні 40 сек., 58°C протягом 1 хв. та за 72°C протягом 45 сек., 30 циклів за 95°C кожні 30 сек., 55°C протягом 1 хв., 72°C кожні 45 сек. на фінальній стадії за 72°C протягом 5 хв. Перші 10 циклів температуру знижували на 0.5°C для того, щоб збільшити резистентність праймерів до мутацій протягом гібридизації. Другий цикл ПЛР включав : 8 хв. денатурації за 95°C, 10 циклів за 95°C кожні 40 сек., 64°C протягом 1 хв. та 72°C протягом 45 сек., 30 циклів за 95°C кожні 30 сек., 59°C протягом 30 сек. та 72°C за 40 сек., і на фінальній стадії за 72°C протягом 5 хв. Для проведення ПЛР пегівірусу типу А (GBVc-NS3) в першому циклі використали праймери GBVc-ak-F1 (5′-CCTTGGACCCAGGTICCNACIGA-3′) та GBVc-ak-R1 (5′-CCTGGTGGGGTRGCIGTNGC-3′) і в другому циклі праймери GBVcak-F2 (5′-GGACCCAGGTGCCIACGGAYGC-3′) та GBVc-ak-R2 (5′-CCTGGTGGGGTRGCGGTNGCRTA-3′). В першому циклі денатурація тривала 8 хв. за 95°C, 10 циклів за 95°C кожні 40 сек., 63°C протягом 1 хв. та 72°C протягом 30 сек., 30 циклів за 95°C кожні 30 сек., 60°C протягом 1 хв., 72°C кожні 40 сек., та на кінцевому етапі за 72°C протягом 5 хв. Перші 10 циклів температуру знижували на 0.5°C для того, щоб збільшити резистентність праймерів до мутацій протягом гібридизації. Протягом другого циклу денатурація тривала 8 хв. за 95°C, 10 циклів за 95°C кожні 40 сек., 71°C протягом 1 хв., і 72°C кожні 30 сек., 30 циклів за 95°C кожні 30 сек., 64°C кожні 30 сек. та 72°C протягом 40 сек., і на фінальній стадії за 72°C протягом 5 хв.
Філогенетичний аналіз та аналіз вторинної структури РНК. Послідовності 5′UTR та ті, що кодують білки, було зіставлено, використовуючи програму MUSCLE, що реалізована в пакеті SSE (37). Було також знайдено розходження сиквенсів та спільні регіони різних геномів та згруповано за допомогою програми Sequence Distance пакету SSE. Нові максимально схожі філогенетичні дерева було побудовано для геліказного регіону NS3 та полімеразного регіону NS5B, використовуючи алгоритм MEGA та оптимальну модель вибрану Model Test. Для обох регіонів було обрано модель Le-Gascuel з гамма розподілом (частота 5) та варіабельністю сайтів (LG + γ + I). Філогенетичний аналіз нуклеотидної послідовності NS3 було представлено за допомогою моделі Jukes-Cantor. РНК структури було передбачено за допомогою Mfold та шляхом пошуку гомологічних структур у різних гепацівірусів. Передбачити псевдо петлю IIIf вірусу гепатиту С та гомологічних структур інших гепацівірусів, використовуючи Mfold та інші алгоритми передбачення вторинних РНК структур не можливо.
Передбачення структур вище, ніж III петля, здійснено за допомогою Mfold. Протеолітичні сайти структурних протеїнів гепегівірусу 1 типу було передбачено, використовуючи програму SignalP version 4.1 (16); це було узгоджено з передбаченням позицій цілого ряду структур. Протеолітичні сайти неструктурних генів було передбачено завдяки ряду сайтів, таких як NS2/NS3, NS3/4A, NS4A/4B, NS4B/5A, та NS5A/5B, які до цього було запропоновано для пегівірусів мавп (18). Гепегівірус 1 типу та інші пегівіруси було проаналізовано на наявність GORS шляхом порівняння енергій для фолдингу конститутивних фрагментів нуклеотидних послідовностей з різними послідовностями за допомогою програми MFED scan пакету SEE (37). Мінімальні енергії фолдингу (MFEs) геномів вірусів гризунів було розраховано за допомогою базового налаштування програми Zipfold. Результати MFE було відображено MFEDs, різниця у відсотках MFE нативної структури від середнього значення відрізняється на 50 (32).
Номер доступу нуклеотидної послідовності. Повний геном гепегівірусу 1 типу зареєстровано в GenBank під номером KT439329.
Список подяки авторам
Статтю було підготовано на основі зразків MHCS та TTVS, отриманих з координаційного центру 9NHLBI Biological Specimen та Data Repository Information Coordinating Center). Дана стаття не містить відгуків MHCS, TTVS, або NHLBI. Ми також вдячні Joel Garcia за підготовку матеріалу для проведення секвенування та Komal Jain і Adrian Caciula за біоінформатичний аналіз. Дослідження фінансувалось грантами NIH HL119485 (A. Kapoor), AI107631 (A. Kapoor) та AI109761 (Lipkin). База даних структур білків (P.D.B.) підтримувалось дослідницькою програмою (Intramural Research Program of National Institute of Dental and Craniofacial Research), NIH.
Джерело: Mbio.asm.org
Переклад з англійської: Еліана Духно
Зі списком використаної авторами літератури можна ознайомитись перейшовши за посиланням на першоджерело; у тексті перекладу посилання на літературні джерела збережено.