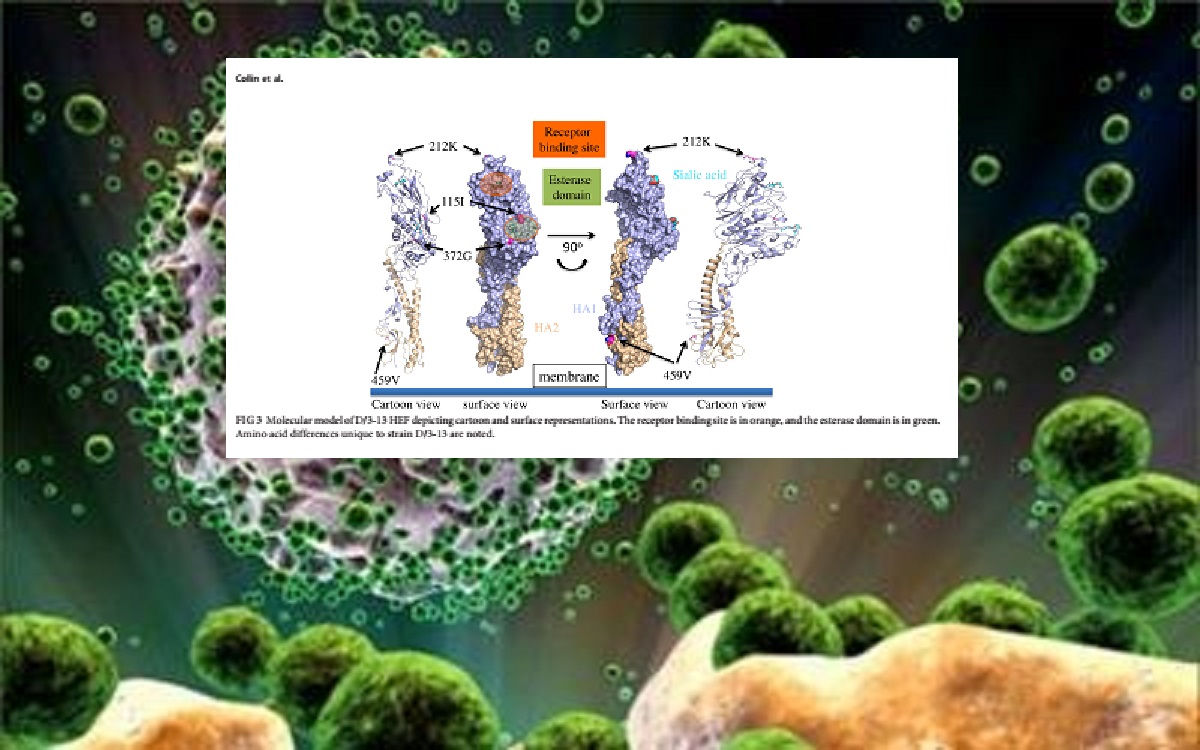
 Viruses with approximately 50% homology to human influenza C virus (ICV) have recently been isolated from swine and cattle. The overall low homology to ICV, lack of antibody cross-reactivity to ICV in hemagglutination inhibition (HI) and agar gel immunodiffusion assays, and inability to productively reassort with ICV led to the proposal that these viruses represented a new genus of influenza virus, influenzavirus D (IDV). To further our understanding of the epidemiology of IDV, real-time reverse transcription-PCR was performed on a set of 208 samples from bovines with respiratory disease. Ten samples (4.8%) were positive and six viruses were successfully isolated in vitro. Phylogenetic analysis of full-genome sequences of these six new viruses and four previously reported viruses revealed two distinct cocirculating lineages represented by D/swine/Oklahoma/1334/2011 (D/OK) and D/bovine/Oklahoma/660/2013 (D/660), which frequently reassorted with one another. Antigenic analysis using the HI assay and lineage-representative D/OK and D/660 antiserum found up to an approximate 10-fold loss in cross-reactivity against heterologous clade antiserum. One isolate, D/bovine/Texas/3-13/2011 (D/3-13), clustered with the D/660 lineage, but also had high HI titers to heterologous (D/OK) clade antiserum. Molecular modeling of the hemagglutinin esterase fusion protein of D/3-13 identified a mutation at position 212 as a possible antigenic determinant responsible for the discrepant HI results. These results suggest that IDV is common in bovines with respiratory disease and that at least two genetic and antigenically distinct clades cocirculate.
Viruses with approximately 50% homology to human influenza C virus (ICV) have recently been isolated from swine and cattle. The overall low homology to ICV, lack of antibody cross-reactivity to ICV in hemagglutination inhibition (HI) and agar gel immunodiffusion assays, and inability to productively reassort with ICV led to the proposal that these viruses represented a new genus of influenza virus, influenzavirus D (IDV). To further our understanding of the epidemiology of IDV, real-time reverse transcription-PCR was performed on a set of 208 samples from bovines with respiratory disease. Ten samples (4.8%) were positive and six viruses were successfully isolated in vitro. Phylogenetic analysis of full-genome sequences of these six new viruses and four previously reported viruses revealed two distinct cocirculating lineages represented by D/swine/Oklahoma/1334/2011 (D/OK) and D/bovine/Oklahoma/660/2013 (D/660), which frequently reassorted with one another. Antigenic analysis using the HI assay and lineage-representative D/OK and D/660 antiserum found up to an approximate 10-fold loss in cross-reactivity against heterologous clade antiserum. One isolate, D/bovine/Texas/3-13/2011 (D/3-13), clustered with the D/660 lineage, but also had high HI titers to heterologous (D/OK) clade antiserum. Molecular modeling of the hemagglutinin esterase fusion protein of D/3-13 identified a mutation at position 212 as a possible antigenic determinant responsible for the discrepant HI results. These results suggest that IDV is common in bovines with respiratory disease and that at least two genetic and antigenically distinct clades cocirculate.
A novel bovine influenza virus was recently identified. Detailed genetic and antigenic studies led to the proposal that this virus represents a new genus of influenza, influenzavirus D (IDV). Here, we show that IDV is common in clinical samples of bovine respiratory disease complex (BRDC), with a prevalence similar to that of other established BRDC etiological agents. These results are in good agreement with the near-ubiquitous seroprevalence of IDV previously found. Phylogenetic analysis of complete genome sequences found evidence for two distinct cocirculating lineages of IDV which freely reassort. Significant antigenic differences, which generally agreed with the surface glycoprotein hemagglutinin esterase phylogeny, were observed between the two lineages. Based on these results, and on the ability of IDV to infect and transmit in multiple mammalian species, additional studies to determine the pathogenic potential of IDV are warranted.
Full text can be downloaded by the clicking on the title:
Cocirculation of Two Distinct Genetic and Antigenic Lineages of Proposed Influenza D Virus in Cattle